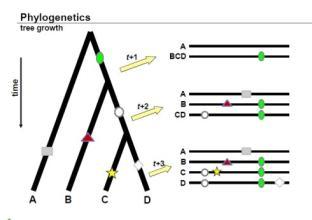
什么叫系统进化?
系统发生分析一般是建立在分子钟基础上的。分子钟:分子序列进化是按照一恒定速率进行的,所以积累突变的数量和进化时间成一定比例,基于这个假说,发生树上的树枝长度可以用来估算基因分离的时间。
什么叫系统进化树(Phyligenetictree)?
系统进化树是对多序列比对(MSA)结果以树形图形式的一个呈现,对于研究进化关系有很大的帮助,通过进化树分析我们也可以关键功能基因和蛋白得出一些假说。
如上图所示,进化树可以有不同的表示形式
上图很好的反映了进化树构建的依据:1,随着物种进化的演绎,进化水平越相近的物种他们的序列越接近;2如果是由同一个物种演化过来的,分化出来的不同物种会保留共同祖先的印记,这是区别于其他的祖先的。
系统进化树分有根(rooted)和无根(unrooted)树。有根树(归于一个节点)反映了树上物种或基因的时间顺序,而无根树只反映分类单元之间的距离而不涉及谁是谁的祖先问题。
进化树的构建
进化树的构建大体要分为3步:序列的比对,建树,然后验证。
1,序列的比对:做ALIGNMENT的软件很多,最经常使用的有CLUSTALX和CLUSTALW
2,构建进化树有两种基本的方法:独立元素法(discrete charactermethods)和距离法(distancemethods),基于距离的构建方法UPGMA(Unweightedpair group method with arithmetic mean,平均连接聚类法)、ME(MinimumEvolution,最小进化法)和NJ(Neighbor-Joining,邻接法);基于特征的构建方法:最大简约法(MP法),最大似然法(ML法),进化简约法(EP法),相容性方法等。
不同的方法可能会得到不同的结论,我们需要用不同的方法以及不同的参数,加上对生物问题的理解来构建最好的进化树来帮助我们更好的理解生物学问题。其中一个衡量树的好坏的方法就是看bootstrap的值,值越大越好,
距离法:
距离依靠法是指进化树的拓扑形状由两两序列的进化距离决定的。进化树枝条的长度代表着进化距离
1,所有的距离法首先通过俩俩比对产生一个“距离矩阵”,然后计算出每对序列的基于距离,简单的理解基因距离就是两个序列没有匹配上的个数(当然,实际计算比这个要麻烦的多);2,然后这个俩俩比对距离矩阵用来判断距离最近的两个序列,这两个序列来形成进化树的两个树枝,这些俩俩比对的距离矩阵然后重新开始找序列最近的两个序列,但这次最相近的两个序列通过一个节点连接到树上,以次往下推,直到结束。3,再根据距离画好这个树。
这个方法的优点就在于快速,缺点为:1它的准确与否是建立在这样的假说上的:additivedistances (always)和molecularclock (sometimes);2Information loss occurs due to data transformation;3 Uninterpretable branch lengths;4Single “best tree” found.
独立元素法:
所谓独立元素法是指进化树的拓扑形状是由序列上的每个碱基/氨基酸的状态决定的(例如:一个序列上可能包含很多的酶切位点,而每个酶切位点的存在与否是由几个碱基的状态决定的,也就是说一个序列碱基的状态决定着它的酶切位点状态,当多个序列进行进化树分析时,进化树的拓扑形状也就由这些碱基的状态决定了)。根据transitionprobabilities, base frequencies, rateheterogeneity等等求出最大的概率图
举个硬币的例子来说明问题吧
Likelihood (L) = Probability(dataobserved | model)
Data :HHTHTH
Model 1: fair coin Prob(H) = 0.5, Prob(T) = 0.5
Model 2: 2-head coin Prob(H) = 1.0, Prob(T) = 0.0
Model 3: 2-tail coin Prob(H) = 0.0, Prob(T) = 1.0
L (Data|Model1)
=Prob(H|Model1) * Prob(H|Model1) * Prob(T|Model1) * Prob(H|Model1)*
Prob(T|Model1) *Prob(H|Model1)
= 0.5 *0.5 * 0.5 * 0.5 * 0.5 * 0.5 = 0.0156
L (Data|Model2) = 1.0 * 1.0 * 0.0 *1.0 * 0.0 * 1.0 = 0.0
L(Data|Model3) = 0.0 *0.0 * 1.0 * 0.0 * 1.0 * 0.0 = 0.0
同理对于maximum likelihood
Find themodel that maximizes the likelihood of the observed data
Data :GGACGCCTGACGCCGCTCGG
Model 1:equal base composition - 0.25, 0.25, 0.25, 0.25 – A, C, G, T,respectively
Model 2:G+C bias - 0.1, 0.4, 0.4, 0.1 – A, C, G, T, respectively
Model 3:A+T bias - 0.4, 0.1, 0.1, 0.4 – A, C, G, T, respectively
L(Data|Model1) = Prob(G|Model1)*Prob(G|Model1)*Prob(A|Model1)*...*Prob(G|Model1) = 0.2520 = 9.1x10-13
L(Data|Model2) = 0.416 * 0.14 = 4.3x10-11 ← maximumlikelihood
L (Data|Model3) = 0.116 * 0.44 =2.6x10-18
对于核酸替代进化模型需要考虑两个因素:1每个碱基出现的概率;2根据进化关系确定的每个碱基转移的概率。有很多进化模型,合理选择。
优点:基于精细的进化模型;可以对特殊的树形拓扑图的相似性统计评估;经常返回很多相似的图(Oftenreturns many equally likely trees),比其他的方法呈现的结果要好。缺点:计算花时间,Often returns many equally likelytrees.
3,Bootstrap验证
这是目前公认的比较好的检验方法,采用随机抽样的方法组成新的序列,然后序列比对,出现同样的比对结果的概率,如下图所示,我们对一定长度序列有放回的抽取,抽取次数跟序列长度一样,pr1为第一次随机抽样的模式,第一个碱基被抽中了1次,第二个碱基被抽中了3次,第三个碱基被抽中了1次……然后以这种模式抽提所有的比对的序列形式新的序列,然后在对新的序列进行构图,同理,随机产生新的模式,重复之前的步骤,这样的随机产生的模式一共1000组,最后求出模中节点出现的概率即为该序列比对的bootstrap概率。
这种方法需要两种假设:数据量足够大;The data are identically andindependently distributed。
Bootstrap values
> 90%strongly supported
70 >90% well supported
50 >70% weakly supported
<50% not supported
上面两个图bootstrap原理一样,但是在建立取点模式上有一点点不一样。
对进化树的分析
假设上面的比对结果都有一个不错的bootstrap验证,基因X’,X’’,X’’’,X’’’’直系同源,Y,Y’跟这几个旁系同源(直系来源于基因的分化,旁系来源于基因的复制),图1是我们的参考进化图,反应的跟我们预期的一样,图2中跟我们预期的图1一比,我们怀疑可能是发生了基因水平转移(HGT);图3,我们发现了一个旁系同源基因,在其他的物种中没有发现旁系同源,所以我们怀疑可能是发生了基因复制;图4我们看到旁系同源基因也跟其他的物种同源,可以推断出这个复制的过程在这两个物种的祖先就已经发生了。
表1 构建分子进化树相关的软件
软件网址说明
ClustalXhttp://bips.u-strasbg.fr/fr/Documentation/ClustalX/图形化的多序列比对工具
ClustalWhttp://www.cf.ac.uk/biosi/resear ...loads/clustalw.html命令行格式的多序列比对工具
GeneDochttp://www.psc.edu/biomed/genedoc/多序列比对结果的美化工具(可以导入fasta格式的文件,出来的图可用于发表,我用过)
BioEdithttp://www.mbio.ncsu.edu/BioEdit/bioedit.html序列分析的综合工具
MEGAhttp://www.megasoftware.net/图形化、集成的进化分析工具,不包括ML
PAUPhttp://paup.csit.fsu.edu/商业软件,集成的进化分析工具
PHYLIPhttp://evolution.genetics.washington.edu/phylip.html免费的、集成的进化分析工具
PHYMLhttp://atgc.lirmm.fr/phyml/最快的ML建树工具
PAMLhttp://abacus.gene.ucl.ac.uk/software/paml.htmlML建树工具
Tree-puzzlehttp://www.tree-puzzle.de/较快的ML建树工具
MrBayeshttp://mrbayes.csit.fsu.edu/基于贝叶斯方法的建树工具
MAC5http://www.agapow.net/software/mac5/基于贝叶斯方法的建树工具
TreeViewhttp://taxonomy.zoology.gla.ac.uk/rod/treeview.html进化树显示工具(加红色标注的为最通用的分析软件)
参考资料: 柳城的博客:http://liucheng.name/577/
BIOINFOMATIC METHOD课程
BaldaufS L. Phylogeny for the faint of heart: a tutorial[J]. TRENDS inGenetics, 2003, 19(6): 345-351.
ps:个人感觉这个的整理非常强大,好多东西都搞明白了