蛋白质纯化技术介绍
蛋白质纯化技术指南
蛋白质的一级、二级、三级和四级结构决定了它的物理、化学、生物化学、物理化学和生物学性质,综述了不同蛋白质之间的性质存在差异或者改变条件是使之具有差异,利用一种同时多种性质差异,在兼顾收率和纯度的情况下,选择蛋白质提纯的方法。
蛋白质在组织或细胞中一般都是以复杂的混合物形式存在,每种类型的细胞都含有成千种不同的蛋白质。蛋白质的分离和提纯工作是一项艰巨而繁重的任务,到目前为止,还没有一个单独的或一套现成的方法能把任何一种蛋白质从复杂的混合物中提取出来,但对任何一种蛋白质都有可能选择一套适当的分离提纯程序来获取高纯度的制品。
蛋白质提纯的总目标是设法增加制品纯度或比活性,对纯化的要求是以合理的效率、速度、收率和纯度,将需要蛋白质从细胞的全部其他成分特别是不想要的杂蛋白中分离出来,同时仍保留有这种多肽的生物学活性和化学完整性。
能从成千上万种蛋白质混合物中纯化出一种蛋白质的原因,是不同的蛋白质在它们的许多物理、化学、物理化学和生物学性质有着极大的不同,这些性质是由于蛋白质的氨基酸的序列和数目不同造成的,连接在多肽主链上氨基酸残基可是荷正电的、荷负电的、极性的或非极性的、亲水的或疏水的,此外多肽可折叠成非常确定的二级结构(α螺旋、β折叠和各种转角)、三级结构和四级结构,形成独特的大小、形状和残基在蛋白质表面的分布状况,利用待分离的蛋白质与其它蛋白质之间在性质的差异,即能设计出一组合理的分级分离步骤。
可依据蛋白质不同性质与之相对应的方法将蛋白质混合物分离:
1.分子大小
不同种类的蛋白质在分子大小方面有一定的差别,可用一些简便的方法,使蛋白质混合物得到初步分离。
1.1透析和超滤
透析在纯化中极为常用,可除去盐类(脱盐及置换缓冲液)、有机溶剂、低分子量的抑制剂等。透析膜的截留分子量为5000左右,如分子量小于10000的酶液就有泄露的危险,在纯化中极为常用,可除去盐类、有机溶剂、低分子量的抑制剂等。超滤一般用于浓缩和脱色
1.2离心分离置换缓冲液
许多酶富集于某一细胞器内,匀浆后离心得得到某一亚细胞成分,使酶富集10~20倍,再对特定的酶进行纯化。差速离心,分辨率较低,仅适用于粗提或浓缩。速率区带法,如离心时间太长所有的物质都会沉淀下来,故需选择最佳分离时间,可得到相当纯的亚细胞成分用于进一步纯化,避免了差速离心中大小组分一起沉淀的问题,但容量较小,只能用于少量制备。等密度梯度离心常用的离主介质有蔗糖、聚蔗糖、氯化铯、溴化钾、碘化钠等等
1.3凝胶过滤
这是根据分子大小分离蛋白质混合物最有效的方法之一,注意使要离的蛋白质分子量落在凝胶的工作范围内。选择不同的分子量凝胶可用于脱盐、置换缓冲液及利用分子量的差异除去热源。
2.形状
蛋白质在离心通过溶液运动时,或通过膜、凝胶过滤填料颗粒或电泳凝胶中的小孔运动时,都会受到形状的影响:对两种相同质量的蛋白质而言,球状蛋白质具有较小的有效半径(斯托克半径),通过溶液沉降时遇到的摩擦力小,沉降较快而显得比其它形状的蛋白质大;反之,在体积排阻色谱时,斯托克半径较小的球状蛋白质更容易扩散进入凝胶过滤填料颗粒内部,较迟洗脱出来,因而显得比其它形状的蛋白质要小。
3.溶解度
利用蛋白质的溶解度的差别来分别各种蛋白质常用的方法。影响蛋白质溶解度的外界因素很多,其中主要有:溶液的pH、离子强度、介电常数和温度,但在同一的特定外界条件下,不同的蛋白质具有不同的溶解度。适当改变外界条件,控制蛋白质混合物中某一成分的溶解度
3.1pH控制和等电点沉淀
蛋白质在其等电点一般较不易溶解。
3.2蛋白质的盐溶和盐析
3.3有机溶剂分级法
蛋白质在不同的溶剂中的溶解度有很大不同,从基本不溶(<10μg/ml)直至极易溶解(>300mg/ml)不等。影响蛋白质溶解度的可变因素包括温度、pH、溶剂的极性、离子性质和离子强度。引起蛋白质沉淀的有机溶剂的浓度不同,故控制有机溶剂的浓度可分离蛋白质。
水溶性非离子聚合物如聚乙二醇也能引起蛋白质的沉淀。
3.4温度
不同的蛋白质在不同的温度具有不同的溶解度和活性。大多数蛋白质在低温下比较稳定,故分离操作一般在0℃或更低温度下进行。
4.电荷
蛋白质净电荷取决于氨基酸残基所带的正负电荷的总和,如中性溶液中带净负电荷则称为酸性蛋白质,
4.1电泳
不仅是分离蛋白质混合物和鉴定蛋白质纯度的重要手段,而且也是研究蛋白质性质很有用的方法。
等电聚焦分辨率很高,pI有0.02pH的差异就能分开。
2D-PAGE分离蛋白质分辨率已经发展到100000个蛋白点。
4.2离子交换层析
改变蛋白质混合物溶液中的盐离子强度、pH和(阴、阳)离子交换填料,不同蛋白质对不同的离子交换填料的吸附容量不同,蛋白质因吸附容量不同或不被吸附而分离。
洗脱可采用保持洗脱剂成分一直不变,也可采用改变洗脱剂的盐度或pH的方法洗脱,后一种可分分段洗脱和梯度洗脱。梯度洗脱一般效果好,分辨率高,特别是使用交换容量小,对盐浓度敏感的离子交换剂,多用梯度洗脱。控制洗脱剂的体积(与柱床体体积相比)、盐浓度和pH,样品组分能从离子交换柱上分别洗脱下来。
蛋白分子暴露在外表面的侧链基团的种类和数量不同,故在一定的PH值和离子强度的缓冲液的所带的电荷不同
5.电荷分布
电荷的氨基酸残基可均匀地分布于蛋白质的表面,既可以适当的强度与阳离子交换柱结合也能以适当强度与阴离子结合,因多数蛋白质都有不能在单一的溶剂条件下同时与两种类型的离子交换柱结合,故可得用此性质纯化;电荷的氨基酸残基亦可成簇分布,使某一区域带强正电荷而另一区域带强负电荷,呈强酸性或强碱性,只能在极端pH与阳离子交换树脂或阴离子交换树脂结合,如钙调蛋白只能在pH2时与阳离子交换树脂结合。
6.疏水性
多数疏水性的氨基酸残基藏在蛋白质的内部,但也有一些在表面。蛋白质表面的疏水性氨基酸残基的数目和空间分布决定了该蛋白质是否具有与疏水柱填料结合从而利用它来进行分离的能力。
因其廉价和纯化后的蛋白质具有生物活性,是一种通用性的分离和纯化蛋白质的工具。高浓度盐水溶液中蛋白质在柱上保留,在低盐或水溶液中蛋白质从柱上被洗脱,故特别适用于浓硫酸铵溶液沉淀分离后的母液以及该沉淀用盐溶解后的含有目标产品的溶液直接进样到柱上,当然也适用7mol/盐酸胍或8mol/L脲的大肠杆菌的治疗蛋白质提取液直接进样到柱上,在分离的同时也进行了复性。
7.密度
多数蛋白质的密度在1.3~1.4g/cm3之间,分级分离蛋白质时一般不常用此性质,不过对含有大量磷酸盐或脂质的蛋白质与一般蛋白质在密度上明显不同,可用密度梯度法离心与大部分蛋白质分离。
8.基因工程构建的纯化标记
通过改变cDNA在被表达的蛋白的氨基端或羧基端加入少许几个额外氨基酸,这个加入的标记可用来作为一个有效的纯化依据。
8.1GST融合载体
使要表达的蛋白质和谷胱甘肽S转移酶一起表达,然后利用Glutathione Sepharose 4B作亲和纯化,再利用凝血酶或因子Xa切开。
8.2蛋白A融合载体
使要表达的蛋白和蛋白A的IgG结合部位融合在一起表达,以IgG Sepharose纯化。
8.3含组氨酸标记(Histidine-tagged)Chelating Sepharose
最通行的标记之一,是在蛋白质的氨基端加上6~10个组氨酸,在一般或变性条件(如8M尿素)下借助它能与Ni2+螯合柱紧紧结合的能力,用咪唑洗脱,或将pH降至5.9使组氨酸充分质子化,不再与结合Ni2+使之得以纯化。
重组蛋白在设计、构建时已融入纯化构想。样品多夹杂了破碎细胞或可溶产物,扩张床吸附技术STREAmlINE适合做粗分离。
9.亲和能力
结合效率高,分离速度快的特点。配基可是酶的底物、抑制剂、辅因子、特异性的抗体、吸附后可改变缓冲液的离子强度和PH的方法,洗耳恭听脱下来,也可用更高浓度的同一配体溶液或亲和力更强的配体溶液洗脱
亲和层析固定相的配基与生物分子之间的特殊的生物大分子亲和能力不同来进行相互分离的,依亲和选择性的高低分为:基团性亲和层析,固定相上的配基对一类基团的极强的亲和力。如含有糖基的一类蛋白质或糖蛋白对三嗪染料显示特别强的吸附能力;高选择性(专一性)亲和层析,配基仅对某一种蛋白质有特别强的亲和性。如单克隆抗体对抗原的特异性的吸附。
亲和层析除特异性的吸附外,仍然会因分子的错误认别和分子间非选择性的作用力而吸附一些杂蛋白质,另洗脱过程中的配体不可避免的脱落进入分离体系。
与超滤结合起来,将两者优点集中形成超滤亲和纯化,具有高分离效率和大规模工业化的优点,适用于初分离。
按配基的不同可分为:
(1)金属螯合介质
过渡金属离子Cu2+、Zn2+和Ni 2+等以亚胺络合物的形式键合到因定相上,由于这些金属离子与色氨酸、组氨酸和半胱氨酸之间形成了配价键,从而形成了亚胺金属—蛋白螯合物,使含有这些氨基酸的蛋白被这种金属螯合亲和色谱的固定相吸附。螯合物的稳定性受单个组氨酸和半胱氨酸解离常数所控制,从而亦受流动相的pH和温度的影响,控制条件可以使不同蛋白质相互分离。
(2)小配体亲和介质
配体有精氨酸、苯甲酰胺、钙调因子、明胶、肝素和赖氨酸等等。
(3)抗体亲和介质
即免疫亲和层析,配体有重组蛋白A和重组蛋白G,但蛋白A比蛋白G专一,蛋白G能结合更多不同源的IgG。
(4)颜料亲和介质
染料层析的效果除主要取决于染料配基与酶的亲和力大小外,还与洗脱缓冲液的种类、离子强度、PH值及待分离的样品的纯度有关。配体有Cibacron Blue和Procion Red两种。在一定的条件下,固定化的染料能起阳离子交换剂的作用,为了避免此现象的发生,最好要离子强度小于0。1和PH大于7时操作。
(5)外源凝集素亲和介质
配体有刀豆球蛋白、扁豆外源凝集素和麦芽外源凝集素,固相外源凝集素能和数种糖类残基发生可逆反应,适合纯化多糖、糖蛋白。
10.非极性基团之间作用力
溶质分子中的非极性基团与非极性固定相间的相互作用力(非选择性分散力或伦敦力)大小与溶质分子极性基团与流动力相中极性分子在相反方向上相互作用力的差异进行分离。因其流动相中的置换剂是极性小于水的有机溶剂(如甲醇、乙腈、四氢呋喃等),这些有机溶剂可能使许多蛋白质分子产生不可逆的变性;流动相中须有离子对试剂(如三氟乙酸、甲酸、磷酸等)存在才可使分离有效地进行和获得高的质量回收率;分离须在酸性介质中进行(一般pH在2~3之间),又有一些蛋白质会在后两种条件下产生不可逆的分子构象变化,故在生物大分子中人分离纯化中受到限制,但分子构象变化可逆的蛋白质而言是有效的方法。
正相色谱在生物大分子中的分离和纯化中应用相对较少,因所用的溶剂很贵。
11.可逆性缔合
在某些溶液条件下,有一些酶能聚合成二聚体、四聚体等,而在另一种条件下则形成单体,如相继在这两种不同的条件下按大小就可以进行分级分离。
12.稳定性
12.1热稳定性
大多数蛋白质加热到95℃时会解折叠或沉淀,利用这一性质,可容易地将一种经这样加热后仍保持其可溶性活性的蛋白质从大部分其它细胞蛋白质中分离开。
12.2蛋白酶解稳定性
用蛋白酶处理上清液,消化杂蛋白,留下抗蛋白酶解抗性蛋白质。
13.分配系数
即利用双水相萃取分离,常用的生物物质分离体系有:聚乙二醇(PEG)/葡聚糖(DEXTRAN)、PEG/磷酸盐、PEG/硫酸铵等。由于具有含水比例高、选用的聚合物及盐对酶无毒性、分离设备与化学工业通用等优点,在工业上目益受重视。
分配行为受聚合物分子大小、成相浓度、PH、无机盐种类等因素影响
发展:具有亲和双水相萃取及膜分离双水相萃取等新型双水相分离技术。
双向水溶液系统的蛋白质纯化
一些与水混合多聚物的不相溶性导致依赖于多聚物浓度的两相系统的液-液分配技术,可以由两不同的高水溶性的多聚物或一种多聚物各一种盐,用于从微生物匀浆中除支细胞碎片、后续分配步骤进一步纯化。分离的选择性一般随着分离分子或颗粒大小面增加。
14.表面活性
14.1泡沫分离
蛋白质溶液具有表面活性,气体在溶液中鼓泡,气泡与液相主体分离,在塔顶富集,达到分离和浓缩的目的。
14.2反胶团相转移法
反胶团相转移法是80年代兴起的一种新型分离技术,它利用表面活性剂分子在有机溶剂中自发形成的反向胶团(反胶团),在一定条件下将水溶性蛋白质分子增溶进反胶团的极性核(水池)中,再创造条件将蛋白质抽提至另一水相,实现蛋白质的相转移,达到分离和提纯蛋白质的目的。反胶团中的蛋白质分子受到周围水分子和表面活性剂极性头的保护,仍保持一定的活性,甚至表现出超活性。由于蛋白质增溶于反胶团与蛋白质所带电荷及反胶团内表面电荷间的静电作用及反胶团的大小有关,因而表面活性剂的种类、水溶液的pH值及离子强度等因素均影响反胶团对蛋白质的相转移。据报道利用AOT/异辛烷反胶团对酵母脂肪酶进行相转移。
14.3聚合物-盐-水液-固苯取体系是90年代在国内开发的种新的萃取体系,成功地用于萃取金属离子及卞果酸脱氢酶等生物活性物质较强的吸附及乳化作用等缺陷,成相容易成相后直接倾出液相即可使液固的相分离,勿需特殊技术处理,不用有机溶剂,无毒性,成相聚合物及盐对生物活性物质有稳定和保护作用,萃取分离选择性好消耗低、易于规模放大的分离生队物活件物质的新技术在聚乙二醇修饰物的聚乙二醇/葡聚糖双水相萃取体系共价作用:主要用于含巯基酶的纯化,通过共价键结合在层析介质上,偶联是可逆的,能还原二硫键的低分子化合物洗脱,如空间位阻,可在含变性剂的缓冲液时吸附,如已含二硫键,则先用还原剂打开。
蛋白质纯化的一般原则及方法选择
蛋白质纯化技术指南
随着分子生物学的发展,越来越多的科研人员熟练掌握了分子生物学的各种试验技术,并研制成套试剂盒,使基因克隆表达变得越来越容易lIl。但分子生物学的上游工作往往并非是最终目的,分子克隆与表达的关键是要拿到纯的表达产物,以研究其生物学作用,或者大量生产出可用于疾病治疗的生物制品。相对与上游工作来说,分子克隆的下游工作显得更难,蛋白纯化工作非常复杂,除了要保证纯度外,蛋白产品还必须保持其生物学活性。纯化工艺必须能够每次都能产生相同数量和质量的蛋白,重复性良好。这就要求应用适应性非常强的方法而不是用能得到纯蛋白的最好方法去纯化蛋白。在实验室条件下的好方法却可能在大规模生产应用中失败,因为后者要求规模化,且在每日的应用中要有很好的重复性。本文综述了蛋白质纯化的基本原则和各种蛋白纯化技术的原理、优点及局限性,以期对蛋白纯化的方法选择及整体方案的制定提供一定的指导。
1 蛋白纯化的一般原则
蛋白纯化要利用不同蛋白间内在的相似性与差异,利用各种蛋白间的相似性来除去非蛋白物质的污染,而利用各蛋白质的差异将目的蛋白从其他蛋白中纯化出来。每种蛋白间的大小、形状、电荷、疏水性、溶解度和生物学活性都会有差异,利用这些差异可将蛋白从混合物如大肠杆菌裂解物中提取出来得到重组蛋白。蛋白的纯化大致分为粗分离阶段和精细纯化阶段二个阶段。粗分离阶段主要将目的蛋白和其他细胞成分如DNA、RNA等分开,由于此时样本体积大、成分杂,要求所用的树脂高容量、高流速,颗粒大、粒径分布宽.并可以迅速将蛋白与污染物分开,防止目的蛋白被降解。精细纯化阶段则需要更高的分辨率,此阶段是要把目的蛋白与那些大小及理化性质接近的蛋白区分开来,要用更小的树脂颗粒以提高分辨常用的离子交换柱和疏水柱,应用时要综合考虑树脂的选择性和柱效两个因素。选择性指树脂与目的蛋白结合的特异性,柱效则是指蛋白的各成分逐个从树脂上集中洗脱的能力,洗脱峰越窄,柱效越好。仅有好的选择性,洗脱峰太宽,蛋白照样不能有效分离。
2.各种蛋白纯化方法及优缺点
2.1蛋白沉淀蛋白能溶于水是因为其表面有亲水性氨基酸。在蛋白质的等电点处若溶液的离子强度特别高或特别低,蛋白则倾向于从溶液中析出。硫酸铵是沉淀蛋白质最常用的盐,因为它在冷的缓冲液中溶解性好,冷的缓冲液有利于保护蛋白的活性。硫酸铵分馏常用做纯化的第一步,它可以初步粗提蛋白质,去除非蛋白成分。蛋白质在硫酸铵沉淀中较稳定,可以短期在这种状态下保存中间产物,当前蛋白质纯化多采用这种办法进行粗分离翻。在规模化生产上硫酸铵沉淀方法仍存在一些问题,硫酸铵对不锈钢器具的腐蚀性很强。其他的盐如硫酸钠不存在这种问题,但其纯化效果不如硫酸铵。除了盐析外蛋白还可以用多聚物如PEG和防冻剂沉淀出来,PEG是一种惰性物质,同硫酸铵一样对蛋白有稳定效果,在缓慢搅拌下逐渐提高冷的蛋白溶液中的PEG浓度,蛋白沉淀可通过离心或过滤获得,蛋白可在这种状态下长期保存而不损坏。蛋白沉淀对蛋白纯化来说并不是多么好的方法,因为它只能达到几倍的纯化效果,而我们在达到目的前需要上千倍的纯化。其好处是可以把蛋白从混杂有蛋白酶和其他有害杂质的培养基及细胞裂解物中解脱出来。
2.2 缓冲液的更换虽然更换缓冲液不能提高蛋白纯度,但它却在蛋白纯化方案中起着极其重要的作用。不同的蛋白纯化方法需要不同pH及不同离子强度的缓冲液。假如你用硫酸铵将蛋白沉淀出来,毫无疑问蛋白是处在高盐环境中,需要想办法脱盐,可用的方法有利用半透膜透析,通过勤换透析液体去除盐分,此法尚可,但需几个小时,通常要过夜,也难以用予大规模纯化中。新型的设备将透析膜夹在两个板中间,板的一侧加缓冲液,另一侧加需脱盐的蛋白溶液,并在蛋白溶液一侧通过泵加压,可以使两侧溶液在数小时内达到平衡,若增加对蛋白溶液的压力,还可迫使水分和盐更多通过透析膜进入透析液达到对蛋白浓缩的目的。也有出售的脱盐柱,柱内的填料是小孔径的颗粒,蛋白分子不能进入孔内,先于高浓度盐离子从柱中流出,从而使二者分离。蛋白纯化的每一步都会造成目的蛋白的丢失,缓冲液平衡的步骤尤甚。蛋白会结合在任何它能接触的表面上,剪切力、起泡沫和离子强度的快速变化很容易让蛋白失活。
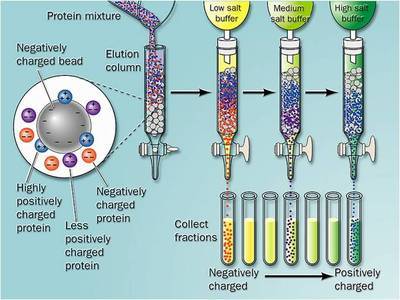
2.3 离子交换色谱这是在所有的蛋白纯化与浓缩方法中最有效方法。基于蛋白与离子交换树脂间的相互电荷作用,通过选择不同的缓冲液,同一种蛋白既可以和阴离子交换树脂(能结合带负电荷的分子)结合,也可以和阳离子交换树脂结合。树脂所用的带电基团有四种:二乙基氨基乙基用于弱的阴离子交换树脂;羧甲基用于弱的阳离子交换树脂;季铵用于强阴离子交换树脂;甲基磺酸酯用于强阳离子交换树脂。蛋白质由氨基酸组成,氨基酸在不同的pH环境中所带总电荷不同。大多数蛋白在生理pH(pH 6—8)下带负电荷,需用阴离子交换柱纯化,极端的pH下蛋白会变性失活.应尽量避免。由于在某个特定的pH下不同的蛋白所带电荷数不同,与树脂的结合力也不同,随着缓冲液中盐浓度的增加或pH的变化,蛋白按结合力的强弱被依次洗脱。在工业化生产中更多地是改变盐浓度而不是去改变pH值,因为前者更容易控制。在实验室中几乎总是用盐浓度梯度去洗脱离子交换柱,利用泵的辅助可以使流入柱的缓冲液中盐浓度平稳地上升,当离子强度能够中和蛋白的电荷时,蛋白就被从柱上洗脱下来。但在工业生产中盐浓度很难精确控制,所以常用分步洗脱而不足连续升高的盐梯度。与排阻层析相比,离子交换特异性更好,有更多的参数可以调整以获得最优的纯化效果,树脂也比较便宜。值得一提的是,即便是用最精确控制的条件,仅用离子交换单一的方法也得不到纯的蛋白,还需要其他的纯化步骤。
2.4 亲和层析亲和层析基于目的蛋白与固相化的配基特异结合而滞留,其他杂蛋白会流过柱子。本方法存在的问题是:单抗非常昂贵,而且也需先纯化;单抗与目的蛋白结合力太强.要用苛刻的条件来洗脱,这会使目的蛋白失活并破坏单抗;混合物中的其他蛋白如蛋白酶也可能破坏抗体或与它们非特异结合;某些单抗也会在纯化过程中从树脂上解离下来混入产物中,也需要从终产物中去除。亲和柱通常在纯化过程的后期应用,此时标本体积已缩小,大部分的杂质已经去除。谷胱甘肽S一转移酶(Glutathione S—transferase,GST)是最常用的亲和层析纯化标签之一,带有此标签的重组蛋白可用交联谷胱甘肽的层析介质纯化,但本方法有以下缺点:首先,蛋白上的GST必须能合适地折叠,形成与谷胱甘肽结合的空间结构才能用此方法纯化;其次,GST标签多达220个氨基酸,如此大的标签可能会影响表达蛋白的可溶性,使形成包涵体,这会破坏蛋白的天然结构,难于进行结构分析,有时即便纯化后再酶切去除GST标签也不一定能解决问题。另一种可应用的亲和纯化标签是6组氨酸标签,组氨酸的咪唑侧链可亲和结合镍、锌和钴等金属离子,在中性和弱碱性条件下带组氨酸标签的目的蛋白与镍柱结合,在低pH下用咪唑竞争洗脱。组氨酸标签与GST相比有许多优点,首先,由于只有6个氨基酸,分子量很小,一般需要酶切去除:其次,可以在变性条件下纯化蛋白,在高浓度的尿素和胍中仍能保持结合力;另外6组氨酸标签无免疫原性,重组蛋白可直接用来注射动物,也不影响免疫学分析。虽然有这么多的优点,但此标签仍有不足,如目的蛋白易形成包涵体、难以溶解、稳定性差及错误折叠等。镍柱纯化时金属镍离子容易脱落漏出混入蛋白溶液,不但会通过氧化破坏目的蛋白的氨基酸侧链,而且柱子也会非特异吸附蛋白质,影响纯化效果。若目的蛋白可与某种碳水化合物特异结合,或者需要某种特殊的辅因子,可将该碳水化合物或辅因子固相化制成亲和柱,结合后目的蛋白可用高浓度的碳水化合物或辅因子洗脱。
2.5 疏水作用层析蛋白是由疏水性和亲水性氨基酸组成’的。疏水性氨基酸位于蛋白空间结构的中心部位,远离表面的水分子。亲水性氨基酸残基则位于蛋白表面。由于亲水性氨基酸吸引了许多的水分子,所以通常情况下整个蛋白分子被水分子包围着,疏水性氨基酸不会暴露在外。在高盐浓度的环境中蛋白的疏水性区域则会暴露并与疏水性介质表面的疏水性配基结合。不同的蛋白疏水性不同,与疏水作用力大小也不同,通过逐渐降低缓冲液中盐浓度冲洗柱子,在盐浓度很低时,蛋白恢复自然状态,疏水作用力减弱被洗脱出来。疏水性树脂的选择性是由疏水性配基的结构决定的,常用的直链配体为烷基配体(alkyl ligands)和芳基配体(arylligands),链越长结合蛋白的能力也越强。理想树脂种类的选择应根据目的蛋白的化学性质而定,不能选择结合力太强的树脂,结合力太强的树脂会很难洗脱,所以开始时应选用中等结合力的苯基树脂探讨条件。为了使选择合适的介质更容易,Amersham Biosciences推出了疏水作用树脂选择试剂盒,里面包括5种不同的树脂供比较。疏水层析很适合作为离子交换纯化的下一个步骤,因为疏水作用层析在高盐浓度下上样,从离子交换得到的产物不需更换缓冲液即可使用。蛋白又在低盐缓冲液中洗脱,又省去了下一步纯化前的更换缓冲液的步骤,既节约了时间,又减少了蛋白的丢失。
2.6 排阻层析也叫凝胶过滤或分子筛。排阻层析柱的填充颗粒是多孔的介质,柱中围绕着颗粒所能容纳的液体量叫流动相,也称无效体积。太大的蛋白不能进入颗粒的孔内,只能存在于无效体积的溶液中,将会最早从柱中洗脱出来,对这部分蛋白无纯化效果。由于各种蛋白的分子大小不同,扩散进入特定大小孔径颗粒内的能力也各异。大的蛋白分子会被先洗脱出来,分子越小,洗脱出来的越晚。为得到最佳的纯化效果,应将孔径大小选在目的蛋白能在无效体积和总柱床体积的中点附近洗脱。排阻层析有其他方法所不具备的优点,首先所能纯化的蛋白分子量范围宽,Tosoh Biosep公司的聚合物树脂,排阻极限可达20o000kD;其次,树脂微孔的形状适合分离球形的蛋白质,纯化过程中也不需要能引起蛋白变性的有机溶剂。应该注意的是某些蛋白不适合用凝胶过滤纯化,因为本技术所用树脂有轻度的亲水性,电荷密度较高的蛋白容易吸附在上面。排阻层析从不用于纯化过程的早期,因为这种方法要求标本高度浓缩,上样量只能在柱体积的1%--4%之间,柱子要细而长才能得到好的分离效果,树脂本身也比较昂贵,规模化的工业生产中不太适用。
2.7 丙烯酰胺凝胶电泳通常用来查看蛋白混合物样品的复杂程度和监测纯化效果。这种方法分离效果极好,可惜很难在不丧失精度情况下放大到制备规模,因为随着胶厚度的增加,电泳时的热效应会严重干扰蛋白的泳动。在基础研究中,有时仅需要少量的纯蛋白进行研究,如蛋白质测序等,此时电泳纯化不失为一种简便快速的好方法。丙烯酰胺凝胶电泳也是蛋白纯化过程中重要的分析工具,可以检测目的蛋白是在哪个梯度的离子交换柱盐洗脱液中;可用来判定近年来随着各学科的迅猛发展,对蛋白纯化技术的需求不断增长,已有的纯化方法被日益改进,新型的纯化方法也相继涌现。羟磷灰石是磷酸钙的结晶,由于其理化性质不够稳定,结合能力差,很难用于层析。进来Bio—Rad公司对其进行了改进,提高了钙和磷的比例,使形成球形、多孔、性质稳定的陶瓷羟磷灰石颗粒,其带正电的钙离子和负电性的磷酸根离子可分别与蛋白的羧基及氨基结合。通过调整缓冲液的pH值,酸性及碱性氨基酸可选择性地与此树脂结合,改变缓冲液的盐浓度可将蛋白洗脱分离。资料显示,使用这种方法能使两种等电点、分子量和疏水性相同的蛋白很好分离。亲和纯化方面,Sigma发展了利用FLAG标签的纯化方法,FLA G序列为N—AspTyrLysAspAspAspAsp-Lys-C,分子量小且亲水性,与其融合表达的蛋白不易形成包涵体,活性也不受影响,用该公司抗FLA G抗体亲和树脂即可纯化目的蛋白。
蛋白质分离纯化的一般程序
蛋白质纯化技术指南
蛋白质分离纯化的一般程序可分为以下几个步骤:
材料的预处理及细胞破碎
分离提纯某一种蛋白质时,首先要把蛋白质从组织或细胞中释放出来并保持原来的天然状态,不丧失活性。所以要采用适当的方法将组织和细胞破碎。常用的破碎组织细胞的方法有:
1.机械破碎法
这种方法是利用机械力的剪切作用,使细胞破碎。常用设备有,高速组织捣碎机、匀浆器、研钵等。
2.渗透破碎法
这种方法是在低渗条件使细胞溶胀而破碎。
3.反复冻融法
生物组织经冻结后,细胞内液结冰膨胀而使细胞胀破。这种方法简单方便,但要注意那些对温度变化敏感的蛋白质不宜采用此法。
4.超声波法
使用超声波震荡器使细胞膜上所受张力不均而使细胞破碎。
5.酶法
如用溶菌酶破坏微生物细胞等。
蛋白质的抽提
通常选择适当的缓冲液溶剂把蛋白质提取出来。抽提所用缓冲液的pH、离子强度、组成成分等条件的选择应根据欲制备的蛋白质的性质而定。如膜蛋白的抽提,抽提缓冲液中一般要加入表面活性剂(十二烷基磺酸钠、tritonX-100 等),使膜结构破坏,利于蛋白质与膜分离。在抽提过程中,应注意温度,避免剧烈搅拌等,以防止蛋白质的变性。
蛋白质粗制品的获得
选用适当的方法将所要的蛋白质与其它杂蛋白分离开来。比较方便的有效方法是根据蛋白质溶解度的差异进行的分离。常用的有下列几种方法:
1.等电点沉淀法
不同蛋白质的等电点不同,可用等电点沉淀法使它们相互分离。
2.盐析法
不同蛋白质盐析所需要的盐饱和度不同,所以可通过调节盐浓度将目的蛋白沉淀析出。被盐析沉淀下来的蛋白质仍保持其天然性质,并能再度溶解而不变性。
3.有机溶剂沉淀法
中性有机溶剂如乙醇、丙酮,它们的介电常数比水低。能使大多数球状蛋白质在水溶液中的溶解度降低,进而从溶液中沉淀出来,因此可用来沉淀蛋白质。此外,有机溶剂会破坏蛋白质表面的水化层,促使蛋白质分子变得不稳定而析出。由于有机溶剂会使蛋白质变性,使用该法时,要注意在低温下操作,选择合适的有机溶剂浓度。
样品的进一步分离纯化
用等电点沉淀法、盐析法所得到的蛋白质一般含有其他蛋白质杂质,须进一步分离提纯才能得到有一定纯度的样品。常用的纯化方法有:凝胶过滤层析、离子交换纤维素层析、亲和层析等等。
有时还需要这几种方法联合使用才能得到较高纯度的蛋白质样品。
蛋白质纯化常用方法
蛋白质纯化技术指南
蛋白质纯化亲和层析法
若表达蛋白质上含有一段六个His 的片段,而亲和吸附胶上接有镍离子,此蛋白质会特异性地结合到吸着胶体;洗去杂质后可imidazole 洗脱目标蛋白质。(Pharmacia 操作手册, Affinity Chromatography)。
仪器设备:
亲和层析管柱 (Bio-Rad 731-1550 Poly-Prep column, 0.8×4 cm)
部分收集器 (另需准备干净试管约25 支)
药品试剂:
金属螯合亲和层析胶体 (Ni-NTA agarose, QIAGEN 30210) 1 mL
◆ 在交联琼脂糖凝胶上接有nitrilotriacetic acid (NTA) 官能基,可以螯合镍离子;使用前先以镍离子饱和的。
Buffer B-300/0 (50 mM Na3PO4, pH 8.0; 0.3 M NaCl)
◆ 使用前才添加成10 mM β-mercaptoethanol.
Buffer B-300/20: Buffer B-300/0 加有20 mM imidazole
Buffer B-300/250: Buffer B-300/0 加有250 mM imidazole
方法步骤:
1) 亲和层析胶体1 mL 已经装填在管柱内,成为一淡蓝色胶柱。请把管柱架直在铁架上,去除下方的填塞物,用buffer B-300/0 流洗20 mL 后塞住出口,准备注入样本。
2) 除去胶面上方的缓冲液,并取出样本 (IEX) 使其回到室温,慢慢用滴管加到亲和胶体上,小心勿弄乱胶体表面;让样本没入胶体中,同时收集流出液每管2.5 mL.
3) 待样本全部进入胶体后,关闭出口,再慢慢加入buffer B-300/20,打开出口收集流出液;buffer B-300/20 共流洗30 mL.
4) 接着以buffer B-300/250 流洗30 mL,方法同上,收集。
5) 所有收集均定量蛋白质并测定酶活性,取收集酶活性最高的数个,保留100 μL 以供分析。
蛋白质纯化离子交换法
各种蛋白质分子上可能带有不同的电性,经过离子交换管柱,可依其分子带电性的差异而分离开来 (Pharmacia 操作手册, Ion Exchange)。
仪器设备:
塑料管柱 (Bio-Rad Econo-Pac column 732-1010, 1.5×12 cm)
部分收集器 (fraction collector, 另需准备干净试管约60 支)
浓缩用离心机 (低速5,000 rpm) 及浓缩离心管Centriplus
药品试剂:
DEAE Sephacel (胶体体积约15 mL,预先以缓冲液Buffer A-0 平衡好)
Buffer A-0 (如上述配法)
Buffer A-300 溶离液:Buffer A-0 加有0.3 M NaCl
Buffer A-500 溶离液:Buffer A-0 加有0.5 M NaCl
方法步骤:
1) 将Econo-Pac 管柱架好,并将三向阀及软管等组合完成。
2) 震荡DEAE Sephacel 胶体使的悬浮,小心倒入管柱中,让胶体慢慢沈降,在沈降过程中随时加入buffer A-0,勿使胶体干掉。
3) 待胶体沈降完全后,高度应在15 cm 左右。胶柱以buffer A-0 一直流洗,流速快慢可以不考虑;连接好收集器,每试管收2.5 mL.离子交换法的胶体平衡极为重要,可测流出液的pH 或离子浓度,看是否与buffer A-0 相同,若不一样则需要再流洗的。
4) 样本的添加方法同上述胶体过滤法,并启动部分收集器开始收集,当样本全部没入胶体后,再加入同体积buffer A-0.以buffer A-0 流洗50 mL 后,去除胶体上方的缓冲液,但勿使胶体干掉。
5) 然后依序以buffer A-300, buffer A-500 溶离的,每批次流洗各50 mL,注意更换浓度时,胶体上方勿残存上一种溶液。
6) 收集后,进行蛋白质定量分析以及GUS 活性测定,结果作图。
7) 收集GUS 活性区,并以Centriprep-30 浓缩,记得要保留100 μL.
8) 离子交换胶体以buffer A-0 洗过50 mL 后,收起来交回。
蛋白质纯化胶体过滤法
不同大小的蛋白质分子进入胶体过滤管柱,可依其分子量差异分离;是一种广泛应用的partition色析法 (Pharmacia操作手册, Gel Filtration)。
仪器设备:色析管柱 (Pharmacia C column, 1.6×100 cm)、铁架、铁夹及水平仪;部分收集器(fraction collector, 需准备干净试管约100 支);浓缩用离心机 (低速5,000 rpm);浓缩用离心管Centriprep-30 (Amicon 4322) 请注意其使用方法
药品试剂:胶体Sephacryl S-300(Pharmacia):a. 预先以缓冲液buffer A-150 平衡好,并且使完全沈降后的胶体体积,占全部体积的七至八成;要先预估好胶体的使用量。b. 胶体温度要与操作场所的温度一致,否则温度变化会产生气泡。c. Sephacryl 系列胶体有相当大的吸附力,因此要在缓冲液加入0.15 M以上的NaCl 以除去非专一性吸附。Buffer A-150:注意使用时的温度要与管柱胶体的温度一致标准分子量组合(Bio-Rad 151-1901):溶于1 mL 后每组取0.4 mL.含有thyroglobulin (670 kD),bovine gamma globulin (158 kD),chicken ovalbumin(44 kD),equine myoglobin (17 kD),vitamin B12(1,350)。
方法步骤:
管柱装填:
1)以纯水冲洗玻璃管柱 (以纯水上下冲洗即可,严禁使用试管刷);并请了解管柱的构造与拆装方法,垂直架好管柱,以软管连接部分收集器,并以bufferA-150 试看管路是否通畅;可以用止血钳或长尾文书夹夹住出口软管,则可控制溶离的进行。 注意系统的摆设要适当,不要装置于交通要冲。
2)依预估量取出Sephacryl 胶体,注意胶体的温度与缓冲液是否已平衡;将瓶中的胶体上下震荡,使的完全悬浮,但勿产生太多气泡。
3)在管柱内加入约10 cm 高缓冲液,然后将胶体慢慢沿着管壁倒入管柱,一直加到管柱顶端,开始流洗后胶体沈降很快。当胶体上方的液面逐渐降低时,可于顶端添加胶体,以达所要高度;胶体高度约90 cm.
4)胶体完全沈降后,小心以buffer A-150 加满管柱,关闭出口,装上顶端端盖并连通缓冲液瓶,打开出口以重力流洗。 调整缓冲液瓶高度,使流速约每五~六秒一滴,并设定收集体积为2.5 mL/tube.
5)胶柱流洗约100 mL 后,关闭出口,拆开顶端端盖,先以滴管吸出胶体上方的溶液到剩约1 cm 高,注意勿破坏胶体表面平整; 然后打开出口,使液面下降至胶体面,再关闭出口,准备注入样本。
样本色析进行:
6)以微量移液器或滴管吸取样本 (样本体积不得超过胶体总体积的3%),沿着胶体上方管壁缓慢加入,注意切勿破坏胶体的平整表面。
7)打开出口,同时开启部分收集器;当样本完全没入胶体时,关闭出口,缓缓加入与样本相等体积的buffer A-150,打开出口待其慢慢进入胶体中,如此重复二次。不得扰动胶体表面,造成凹陷。
8)暂时关闭出口,将液面高度加满至管柱顶端,并把顶端端盖锁上;然后打开出口开始溶离,调整缓冲液瓶的高度,使流速为6 s 一滴。
9)要留心观察前面几个分划,确定整个系统运转无碍,小心部分收集器最容易出问题。管柱预计将流洗过夜,收集约80 管。
10)收集试管,进行蛋白质定量分析以及GUS 活性测定,并请作图。
11)收集GUS 活性区,以Centriprep-30 浓缩至10 mL 后,加buffer A-0 稀释至20 mL,再次浓缩,保留100 μL。
12)管柱请再以buffer A-150 流洗100 mL 后,小心放置一旁,准备以后进行分子量测定。
分子量测定:
13)进行分子量测定前一天,请先以buffer A-150 流洗100 mL,并检查胶柱内有无气泡产生,若有严重的气泡或干裂,必须重新装填管柱。
14)取标准分子量溶液0.4 mL,加上纯质目标酶0.5 mL (以亲和层析法所得的AF 部份),如上法注入管柱中,立刻开始进行胶体过滤,并收集各分划。请依循上述所有管柱及分划收集器的操作要点。
15)收集所得,进行蛋白质定量分析,可定出数个蛋白质尖峰,以作为分子量依据;另以目测法,决定红色高峰的管数,则可定出vitamin B12的溶离管数。利用以上数据,可画出分子量与溶离管数间的直线关系,作为分子量判定的标准校正线。
16)同样的一批分划,请进行酶活性分析(GUS),则可定出酶的溶离体积,对照上述标准校正线,则可求出酶的分子量。
拆除管柱及保存胶体:
17)若管柱长期不用,应当自管柱中取出胶体,以缓冲液清洗后,置冷藏室中保存,但绝对不要放在冷冻箱中。胶体若装填太紧,有时可能不易取出,要有耐心地以缓冲液慢慢冲出来。
18)胶体可以加0.01% NaN3防止霉菌生长,但使用前记得要洗去;再度使用时,请检查胶体中有无灰黑色霉菌颗粒,若有结块而不易打散者,也不要使用。
技术支持
蛋白质纯化技术指南
美国GeneCopoeia公司及其合资企业广州复能基因有限公司的主要业务是生产和销售人类编码全长蛋白的ORF穿梭克隆(Shuttle Clones) 和表达克隆(Expression Clones)。公司利用改进的Gateway?;克隆技术构建了一套20,000多个编码全长蛋白的ORF穿梭克隆。这些ORF穿梭克隆使ORF在不同表达载体间的转换变得简单和快速。公司还进一步地应用拥有自主知识产权的专利技术(RecJoin?;克隆技术)构建了多套编码全长蛋白的ORF表达克隆。这些表达克隆含有不同启动子和不同标签蛋白,适合用于不同表达宿主和无细胞转录-翻译偶联体系。这些表达克隆可直接应用于蛋白质表达、纯化和功能分析而无需进行繁琐的克隆过程。这些编码全长蛋白的ORF来自于经过序列验证的全长cDNA克隆和高质量人体组织的cDNA文库,并通过高保真的克隆技术将其克隆到多套载体中。