线粒体脑肌病伴乳酸酸中毒和卒中样发作综合征
线粒体病是一组线粒体基因或细胞核基因突变导致线粒体结构和功能异常,并由此引起多系统损害的疾病,以需氧量较高的脑和肌肉受累为主[1,2]。其临床表现多样,主要有线粒体脑肌病伴乳酸酸中毒和卒中样发作(Mitochondrialencephalomyopathy with lactic acidosis and stroke-likeepisodes,MELAS)综合征、慢性进行性眼外肌麻痹(CPEO)、肌阵挛性癫痫伴肌肉破碎红纤维(MERRF)综合征、亚急性坏死性脑脊髓病(Leigh病)和以眼外肌麻痹、视网膜色素变性和心脏传导阻滞为特点的Kearns-Sayre综合征(KSS)等多个亚型。MELAS综合征是最常见的一种类型,以线粒体肌病、脑病、乳酸酸中毒和反复卒中样脑部损害为临床表现,于1984年由Pavlakis[3]首先报道。现对MELAS综合征综述如下。
1 遗传学特点
线粒体普遍存在于真核细胞胞质中,是氧化和能量转换的重要场所,亦是细胞核外唯一带有遗传物质的细胞器。线粒体DNA(mtDNA)为双链闭合环状结构,外环为重链(H),内环为轻链(L),由16,569个碱基(bp)组成,含有37个编码基因,分别编码22种tRNA、2种rRNA(12s和16srRNA)以及13种参与氧化磷酸化(OXPHOS)能量产生、呼吸链电子传递过程的蛋白亚单位[4]。MtDNA与核DNA遗传机制不同。其分子遗传特征主要表现为:(1)半自主性:mtDNA是独立于细胞核染色体外的基因组,具有自我复制、转录和翻译的功能。(2)母系遗传:这是线粒体遗传的最突出特点,即母亲将缺陷传递给子女,而只有女儿能将缺陷传递给下一代。这是因为在受精过程中,精子所含的极少的线粒体并不进入卵细胞内,受精卵胞质几乎全部来自卵子,整套线粒体来自母方。在遗传过程中受瓶颈效应(bottleneckeffect)影响,即mtDNA在由母方向子代传递过程中有一个大幅度削减,使子代细胞和组织中仅含有少数卵母细胞的mtDNA。因此,同一家系不同成员间突变比例和临床表型存在很大差异。(3)遗传密码与通用遗传密码不同。(4)异质性和遗传漂变现象:一个细胞中有成百上千个线粒体,一个线粒体中含2~10个DNA分子。当正常和突变的mtDNA以不同比例共存于同一线粒体、细胞、组织或者个体时,称为异质性(heteroplasmy)。异质细胞分裂时,mtDNA通过母系遗传随机分配给子代,由于突变型和野生型mtDNA比例不同,子代的临床表现不一致。经过多代传递,mtDNA表型向野生型或突变型mtDNA占优势方向漂变,异质细胞逐渐向全突变型发展,突变负荷随时间增加。(5)阈值效应:mtDNA表型表达具有阈值效应,即突变mtDNA比例达到一定程度才引起组织、器官功能异常,且症状严重程度取决于突变性质、突变比例及各器官对能量的需求,所以耗能多的器官如肌肉、心脏、脑组织首先受累。(6)高突变率:由于mtDNA没有内含子、保护性组蛋白和缺乏完整有效的修复体系,使之易受氧化磷酸化过程产生的氧自由基的影响,诱发突变几率较核DNA高得多。MELAS综合征相关突变已报道很多,主要位于编码tRNA的基因上,尤其是tRNALeu(UUR)基因,目前报道有7个致病突变位于该基因上(www.mitomap.org)。MELAS相关突变有tRNA编码亚单位上的A3243G/T、G3244A、A3252G、C3256T、T3258C、T3271C、T3291C突变,ND4编码亚单位上的A11084G突变[5],ND5亚单位上G13513A、A13514G、A12770G、A13045C、A13849C[6-9]以及COX3上T9957C突变[9]等。流行病学研究显示儿童中携带A3243G突变比率较高,患病率最低估计约18.4/10万人,但发病率相对低[10]。mtDNA基因tRNALeu(UUR)A3243G点突变是MELAS最常见的致病性突变,约80%的MELAS患者系突变所致,10%左右的患者由T3271C突变引起[11]。A3243G位于tRNALeu(UUR)编码区,3243位点正常A碱基被G碱基替代,致转录终止,妨碍rRNA正常表达和线粒体蛋白质合成,导致呼吸链复合体Ⅰ、Ⅳ活性缺陷,ATP产生不足,从而导致多系统损害。一个基因突变可以引起多种表型或者同一种表型可能由不同突变引起,如携带A3243G突变的患者除表现为典型的MELAS综合征外,还可以表现为其他脑病如CPEO、MERRF、单纯肌病、单纯2型糖尿病伴或不伴神经性耳聋;糖尿病人群中约0.5%~2.8%患者携带A3243G突变[12];Harrison-Gomez等[13]报道一例A3243G突变患者同时累及神经系统、心血管系统和内分泌系统;携带T3271C突变的患者儿童时期症状较轻,至快速终末期出现类似脑炎症状[14];tRNALeu(UUR)基因上G5591A突变则只表现为单纯肌病[15]。有研究显示外周血A3243G突变比例与发病和就诊年龄呈负相关,与病程未见关联[16]。目前线粒体病基因型和表型之间的相关性尚不明确,研究mtDNA突变与临床表型的相关性具有重要意义。
2 临床表现
线粒体病累及多个系统,以能量需求高的组织和器官受累为主,如脑、骨骼肌和心肌。目前,突变基因型和表型之间的关系尚不明确,相同突变可出现不同的临床表现,不同突变亦可产生类似或不同的临床表型,故线粒体病的临床表现具有多样性,且各种亚型的症状可以相互重叠。线粒体功能障碍主要表现为进行性肌肉无力、劳累性肌痛,眼外肌麻痹,中枢和周围神经系统功能障碍以及继发于周期性乳酸酸中毒的一系列症状;其他如心脏、肝脏、肾脏、内分泌系统、血液系统等多个器官系统也可受累。线粒体病遗传特征决定了该病的临床表现具有以下特点:母系遗传;儿童、青少年好发,多在45岁以前发病;脑和骨骼肌等能量需求高的器官容易同时受累;多伴体格和智能发育迟滞,可合并其他系统症状,常合并糖尿病、神经性耳聋;进行性病程,可有不同程度缓解、复发。MELAS早期阶段,患者可表现为肌无力、活动不耐受、身材矮小、癫痫发作、偏头痛、轻偏瘫、偏盲或皮质盲[3],偏头痛和癫痫发作被认为是卒中样发作的最常见症状[17]。典型的MELAS综合征表现包括卒中样发作、脑病、癫痫、偏头痛、智力发育迟滞、肌无力和体型矮小,还可有肌阵挛、共济失调表现,有些患者还伴有听力损害、糖尿病、周围神经病、视神经萎缩、色素性视网膜病、进行性眼外肌麻痹或者心肌病等[18]。心脏受累可表现为心脏扩大、非对称性室间隔肥厚、左心室运动功能弥漫性减退、左心室肥大伴或不伴异常室壁运动或扩张性心肌病等[19]。有些病例以反复发热、头痛、呕吐起病,易误诊为病毒性脑炎。
3实验室检查
3.1生化检查尽管患者临床表现各异,但几乎所有患者均伴有乳酸酸中毒,可以通过测定血清和脑脊液中乳酸、丙酮酸水平作为辅助诊断。疲劳试验可作为简便易行的筛选试验。对MELAS筛查可以采用最小运动量试验,测定运动前后血清乳酸、丙酮酸水平,以乳酸/丙酮酸比值作为判定标准,以运动前血清乳酸/丙酮酸比值>17或者<7、运动后乳酸/丙酮酸比值>22或者<7为异常的标准。血清乳酸水平>0.25g/L,或乳酸/丙酮酸比值>20时,强烈提示线粒体功能障碍;通常情况下脑脊液乳酸值低于血清值,脑脊液中乳酸水平升高,多在0.30~0.40g/L(超过0.20g/L为异常),常提示中枢神经系统受累[20]。如果运动试验中乳酸和丙酮酸同时增高或者两者比值异常,则认为可能存在线粒体肌病。由于该试验只能反映能量代谢和线粒体功能状态,临床上只能作为筛选,不能区分原发性和继发性线粒体肌病。对幼儿或瘫痪病人可采用葡糖糖刺激试验,口服葡萄糖2g/kg后90分钟,对比试验前后乳酸水平,升高2倍以上为异常[21]。其他生化检查亦可发现肌酶、血糖水平升高,线粒体呼吸链复合酶活性降低等。
3.2基因检查mtDNA基因突变分析是诊断该病的最敏感方法。常用的检测方法有聚合酶链反应-限制性片段长度多态性(PCR-RFLP)分析或直接测序,用于对常见突变如A3243G、T3271C、A8344G、T8993G/C等的检测。A3243G、T3271C被认为是MELAS相关突变,A8344G是MERRF相关突变,而T8993G/C则是Leigh综合征相关突变。由于突变异质性,在有丝分裂旺盛的细胞如外周血细胞中检出率低,易出现假阴性,而在有丝分裂不旺盛的肌肉或脑组织中则检出率高。一项对MELAS患者的纵向研究显示突变mtDNA随着血液细胞快速分裂而缓慢被选择,9~19年后突变率降低12%~29%[22]。血液中突变比例相同的患者,临床表现也可截然不同,甚至毫无症状,故血液中突变比例不能很好地反映疾病的严重程度。骨骼肌病检显示RRF和SDH反应增强血管中突变mtDNA比例明显高于非RRF及SDH反应阴性血管[23]。患者肌肉中突变mtDNA比例通常高于外周血。检出率较高的组织为骨骼肌,其次为毛囊、颊粘膜、外周血粒细胞。对线粒体病未知突变进行筛查时,至少应包含常见的突变如A3243G、T3271C、A8344G、T8993G/C等。
3.3神经电生理检查肌电图显示急性去神经支配和肌病表现,多数为肌源性损害,少数可见神经源性改变或两者皆有[21];脑电图检查显示不同程度的弥漫性慢波,正常的α节律减少或消失,可见癫痫脑电图特有的棘慢波、尖慢波综合,具有一定诊断意义[24];各种诱发电位检查对该病也有辅助诊断作用。
4 病理学改变
MELAS综合征的脑组织病理改变为大脑皮质广泛层状坏死或海绵状改变,以颞顶枕叶受损为主,皮质下、深部白质镜下见髓磷脂缺失和神经胶质增生,神经元变性、减少和脱失,小血管增生显著,这种改变在CT上表现为低密度改变,MRIT2-加权则表现为高信号[25,26],亦可见脑皮层、小脑萎缩和基底节区钙化或铁沉积。肌肉活检是诊断线粒体病的重要手段。活检肌肉组织常用的染色方法有改良Gomori三色法(MGT)、琥珀酸脱氢酶(SDH)、还原性辅酶Ⅰ四唑氮还原酶(NADH-TR)、细胞色素C氧化酶(COX)、高碘酸Schiff反应(PAS)、油红O(ORO)染色法等。肌活检组织在光镜下见破碎红纤维(RRF)、SDH反应增强的血管以及受累肌纤维COX缺陷,油红O染色有时可见脂质沉积;电镜下见肌膜下线粒体增多,形态异常和线粒体晶格样包涵体,SDH反应增强的血管平滑肌细胞内线粒体膨胀、异常增生、线粒体嵴减少,部分病例可见脂滴沉积和糖原颗粒增多[27]。RRF、SDH反应增强血管以及电镜下见异常增生线粒体是诊断线粒体肌病的重要病理学依据。RRF是功能异常的大量线粒体在肌膜下聚集形成的,被认为是MELAS综合征特征性病理改变,但并非所有患者均出现,对诊断线粒体脑肌病并无特异性,还可见于强直性肌营养不良、代谢中毒性肌病、包涵体肌炎以及50岁以上的健康老年人[20]。尽管如此,特征性的RRF出现对MELAS的诊断具有重要病理意义,提示电子传递链异常[3]。87.5%患者肌活检光镜下显示血管壁SDH染色反应增强[28],该病理特征在MELAS和MERRF患者的肌肉活检中比较常见,与RRF具有同等的病理诊断意义。检测呼吸链酶复合体活性,可见肌肉或脑组织中线粒体内与呼吸链和细胞氧化有关的酶的活力不同程度下降或缺陷,如复合体Ⅰ、复合体Ⅳ、NADH脱氢酶、细胞色素C氧化酶活性降低[28]。此外,腓肠神经活检可见有髓神经纤维髓鞘肿胀,电镜下显示髓鞘增厚、肿胀,洋葱皮样结构消失呈无定型物质[29]。
5影像学表现
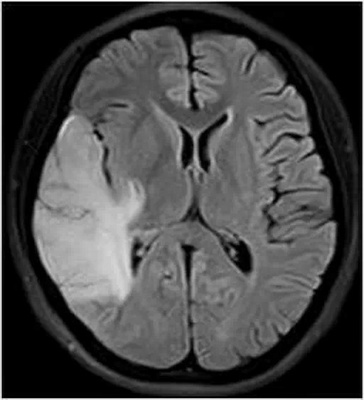
5.1 CTMELAS综合征患者颅脑CT表现为颅内多个梗死样低密度病灶,病变多位于大脑后部颞顶枕叶皮质或皮质下区域,病灶分布与动脉供血区域不一致;急性期可见脑回肿胀和占位效应;基底节区、丘脑、苍白球、壳核、尾状核可见异常钙化灶。
5.2 MRI典型的表现为梗死样病灶,呈长T1、长T2信号,病变主要位于半球后部即颞、顶、枕叶极其交界区皮质或皮质下区域,灰白质均可受累,白质受累轻,有时累及深部白质,酷似分水岭脑梗死,病灶无明显强化或强化不明显,且病灶分布常跨越多个供血区,与动脉供血区不一致,可有脑室扩大及与年龄不相符的脑皮质萎缩,亦可有小脑萎缩改变[30-32]。Scaglia等[31]的研究显示,神经肌肉受累的患者MRI表现为小脑萎缩应考虑线粒体脑肌病的可能。梗死样病灶呈迁移性[32],即一个部位旧的病灶消失,另一个部位新的病灶出现。由于MELAS为发作性疾病,因此在CT或MRI上有病灶反复出现和消退的动态改变。基底区短T1、短T2信号常提示钙化灶存在。病变范围在FLAIR序列上显示较T1WI、T2WI更清楚,呈现高信号,可能由于FLAIR序列对皮层及皮层下病变水肿较敏感和对游离水的抑制,从而体现出其在显示病变范围上的优势。扩散加权成像(DWI)对诊断MELAS比常规MRI更为敏感,急性期病变在DWI上呈现高或稍高信号改变。急性期由于乳酸血症导致血管舒张,病变区呈高灌注和血管源性水肿,此时DWI较常规MRI能更敏感早期地显示该病理改变,此外还可以通过观察表观扩散系数(ADC)是否增高来区分细胞内、外水肿。发病48小时内出现急性神经功能障碍的线粒体脑肌病患者,常规MRI检查可见梗死样病变,其ADC可正常或增高,缺少被阻扩散,而急性卒中患者ADC降低,此表现可与急性脑梗死相鉴别[33]。Iizuka等[17,34]提出的非缺血性神经血管细胞损害学说,可以很好地解释上述神经影像学特点。
5.3 磁共振波谱分析(MRS)MRS可以对MELAS患者脑中多种代谢物进行定量分析,包括乳酸(Lac)、乙酰天门冬氨酸(NAA)、胆碱(Cho)、肌酸(Cr)、肌醇和谷氨酸盐等。正常人的MRS可以观察到4个主要共振波峰,即NAA、Cho、Cr和Lac。Lac是葡萄糖无氧酵解的产物,Lac峰出现常提示组织缺血、缺氧或占位性病变等。典型的MELAS患者大脑卒中样病灶区MRS显示明显的乳酸升高,NAA、Cr、肌醇和谷氨酸水平降低[35]。这种乳酸升高在不同的回波序列上表现不同峰型,在短回波时间(如TE=35 ms)序列的波谱上为正立的双峰,在长回波时间(如TE = 144 ms)序列的波谱上则为倒置的双峰,在更长回波时间(如 TE =288ms)序列的波谱上又为正立的双峰。Abe[36]报道1例患者卒中样发作48小时脑部MRI和DWI均未显示异常信号,而MRS检测出乳酸峰,2周后该部位的DWI显示高信号。这提示MRS有助于MELAS的早期诊断。脑脊液乳酸水平检测为有创检查,不利于重复检测,而MRS在检测中枢神经系统乳酸水平无创且可能更为敏感。通常脑代谢物浓度>1mmol/L,MRS即可检出[37]。Cross等[38]研究结果显示脑脊液乳酸水平>4mmol/L时,1H-MRS能显示稳定的乳酸双峰。说明MRS具有代替传统脑脊液乳酸水平测定的潜能,能无创性地监测患者脑内乳酸的动态变化。MRS检测出乳酸双峰是MELAS的一个特征性表现,用于评价患者脑缺氧的严重程度,但其他病理状态如早期脑梗死、脱髓鞘病变、脑肿瘤等也可以出现,故应结合临床表现综合判断。
5.4 脑血管检查MRA或DSA检查通常无明显血管狭窄征象,但可见病灶分布区血流加快,小血管增多、增粗,这可能源于急性期乳酸血症导致局部血管舒张,病灶呈高灌注和血管源性水肿。慢性期由于能量供应不足导致细胞毒性水肿,影像学改变主要为多发软化灶和皮层萎缩,病灶周围小血管增多和胶质细胞增生,而大血管一般不受累。这点可与缺血性卒中相鉴别。单光子发射断层扫描(SPECT)检查可显示病灶区放射性示踪元素摄取降低,局部脑血流量(rCBF)相对正常,部分区域出现充血状态[39]。质子发射断层扫描(PET)显示病灶区脑血流量正常或轻度增加,葡萄糖代谢率增加,而氧代谢率明显降低[40]。
6 诊断及鉴别诊断
结合MELAS综合征患者临床、生化、病理、影像学和遗传学特点,其诊断依据主要包括:(1)发病年龄一般10~40岁,多为母系遗传,少数散发;(2)临床表现为肌肉无力、运动不耐受、肌萎缩等肌肉受累表现,肌电图多为肌源性改变;发作性头痛、呕吐、癫痫发作、偏盲、偏瘫、精神症状、痴呆等中枢神经系统症状,可伴神经性耳聋、糖尿病、部分眼外肌麻痹等;患者身材矮小,低体重,体质差;(3)运动前后血乳酸、丙酮酸水平升高,肌酶及血糖亦可增高;(4)脑CT及MRI检查显示双侧基底节区钙化,位于半球后部颞、顶、枕叶脑皮质或皮质下区多发卒中样病灶,病灶与血管分布不一致,且随病情发展呈迁移性改变;MRS检查可见乳酸双峰;(5)肌活检可见RRF,电镜下见线粒体增生、形态异常及晶格状包涵体;脑组织病检显示皮质层状或灶状坏死和海棉样改变,胶质细胞增生,小血管弥漫增生;生化测定线粒体功能缺陷;(6)基因检测有mtDNA异常,如A3243G或T3271C突变则更支持诊断。临床上应与脑梗死、病毒性脑炎、重症肌无力、多发性肌炎、进行性肌营养不良等疾病相鉴别。
7 治疗
目前对该病尚无特效治疗,主要是对症支持治疗。
7.1 药物治疗
7.1.1 非特异性治疗(1)能量替代治疗,补充大量三磷酸腺苷(ATP);(2)清除氧自由基,使用抗氧化剂;(3)补充辅酶或辅因子;(4)通过旁路传递电子[41]。目前常用的治疗线粒体病的药物有:辅酶Q10、艾地苯醌、维生素C、维生素E、硫辛酸、硫胺素(维生素B1)、核黄素(维生素B2)、烟酸、维生素K、肌酸、肉碱、二氯乙酸和二甲基甘氨酸等[42,43]。辅酶Q10在线粒体能量生成中具有重要功能,也是有效地抗氧化剂,大量的辅酶Q10可以降低乳酸、丙酮酸水平,改善和缓解症状,临床治疗试验最多。肌酸在MELAS治疗中具有神经保护作用,能够改善线粒体有氧代谢功能,但对合并肾病的患者应慎用[44]。二氯乙酸通过抑制丙酮酸脱氢酶复合体而减少丙酮酸的积聚和乳酸的产生,增加大脑氧代谢和改善脑功能[45]。大量的辅酶Q10、艾地苯醌、肌酸、硫辛酸、维生素B1、维生素B2、细胞色素C等的联合应用可以有效改善症状,提高患者生活质量,控制病情发展。有些研究报道显示这些药物可以明显改善患者肌力,降低运动后乳酸,改善患者生活质量,有些研究则未显示明显的治疗效果,目前尚没有很明确的证据支持这些药物的使用,这些药物的治疗作用还需进一步研究[43]。亦有研究表明丁基苯酞(NBP)能改善由损伤引起的神经细胞线粒体膜电位、膜流动性及呼吸链复合酶IV活性的降低,通过保护线粒体功能、清除氧自由基等机制对低糖低氧损伤导致的神经细胞坏死和凋亡具有良好的保护作用[46],有可能应用于本病治疗。
7.1.2 特异治疗对癫痫、卒中样发作、头痛、肌张力障碍等神经系统症状和非神经系统表现如心脏病、糖尿病等予以对症处理。那些在线粒体功能障碍患者身上更易产生副作用的药物,如甾体类、他汀类、贝特类、异丙酚、神经松弛剂和抗病毒药物等,应慎用或禁用[41]。
7.2 运动疗法(1)耐力训练:Cejudo等[47]的研究显示规律的有氧耐力训练可以改善患者症状,使患者肌肉最大摄氧能力提高28.5%,周围肌力提高32%~62%。有氧耐力训练可以改善患者的生理、生化指标和生活质量,但突变体mtDNA增加可能抵消这些效果,使患者远期预后恶化[48]。(2)阻抗训练:该训练可以使骨骼肌星形细胞转化为成熟的肌纤维,或融入成熟肌纤维内,降低突变负荷[48]。
7.3 饮食疗法:高蛋白、高碳水化合物、低脂饮食能减少脂肪分解,降低糖异生。
7.4 其他治疗对心脏严重受累的患者,可植入心脏起搏器、置入心律转复除颤器或支架等。关于基因治疗的研究很多,但目前主要停留在细胞和动物试验阶段,成功的线粒体基因治疗策略尚未实现[49]。
MELAS综合征是线粒体脑肌病的最常见类型,其临床表现多样性,容易误诊和漏诊。对MELAS的诊断需结合临床表现,通过血清乳酸、丙酮酸最小运动量试验作筛选,影像学检查作辅助,以肌肉活检或线粒体DNA突变检测进一步明确诊断。目前尚无根治性治疗方法,主要为对症支持治疗,及早和有效的个体治疗可以改善患者的生活质量,延缓疾病进展,改善预后。
References
[1]黄光, 王俊芳, 唐煜, 邹凤军. 继发于肺栓塞的全身多器官栓塞1例报告. 临床神经病学杂志. 2011. (01): 37.
[2]白亚辉, 任传成. 线粒体DNA突变与线粒体脑肌病. 临床神经病学杂志. 2008. (06): 471-472.
[3]Pavlakis SG, Phillips PC, DiMauro S, De Vivo DC, Rowland LP.Mitochondrial myopathy, encephalopathy, lactic acidosis, andstrokelike episodes: a distinctive clinical syndrome. Ann Neurol.1984. 16(4): 481-8.
[4]DiMauro S, Schon EA. Mitochondrial respiratory-chain diseases. NEngl J Med. 2003. 348(26): 2656-68.
[5]Lertrit P, Noer AS, Jean-Francois MJ, et al. A new disease-relatedmutation for mitochondrial encephalopathy lactic acidosis andstrokelike episodes (MELAS) syndrome affects the ND4 subunit of therespiratory complex I. Am J Hum Genet. 1992. 51(3): 457-68.
[6]Pulkes T, Eunson L, Patterson V, et al. The mitochondrial DNAG13513A transition in ND5 is associated with a LHON/MELAS overlapsyndrome and may be a frequent cause of MELAS. Ann Neurol. 1999.46(6): 916-9.
[7]Corona P, Antozzi C, Carrara F, et al. A novel mtDNA mutation inthe ND5 subunit of complex I in two MELAS patients. Ann Neurol.2001. 49(1): 106-10.
[8]Liolitsa D, Rahman S, Benton S, Carr LJ, Hanna MG. Is themitochondrial complex I ND5 gene a hot-spot for MELAS causingmutations. Ann Neurol. 2003. 53(1): 128-32.
[9]Choi BO, Hwang JH, Kim J, et al. A MELAS syndrome family harboringtwo mutations in mitochondrial genome. Exp Mol Med. 2008. 40(3):354-60.
[10]Uusimaa J, Moilanen JS, Vainionpaa L, et al. Prevalence,segregation, and phenotype of the mitochondrial DNA3243A>G mutation in children. Ann Neurol. 2007.62(3): 278-87.
[11]Yasukawa T, Suzuki T, Ueda T, Ohta S, Watanabe K. Modificationdefect at anticodon wobble nucleotide of mitochondrialtRNAs(Leu)(UUR) with pathogenic mutations of mitochondrialmyopathy, encephalopathy, lactic acidosis, and stroke-likeepisodes. J Biol Chem. 2000. 275(6): 4251-7.
[12]Suzuki S, Oka Y, Kadowaki T, et al. Clinical features of diabetesmellitus with the mitochondrial DNA 3243 (A-G) mutation inJapanese: maternal inheritance and mitochondria-relatedcomplications. Diabetes Res Clin Pract. 2003. 59(3): 207-17.
[13]Harrison-Gomez C, Harrison-Ragle A, Macias-Hernandez A,Guerrero-Sanchez V. [A3243G mitochondrial DNA mutation andheterogeneous phenotypic expression]. Rev Med Inst Mex Seguro Soc.2009. 47(2): 219-25.
[14]Stenqvist L, Paetau A, Valanne L, Suomalainen A, Pihko H. Ajuvenile case of MELAS with T3271C mitochondrial DNA mutation.Pediatr Res. 2005. 58(2): 258-62.
[15]Swalwell H, Deschauer M, Hartl H, et al. Pure myopathy associatedwith a novel mitochondrial tRNA gene mutation. Neurology. 2006.66(3): 447-9.
[16]方方, 马袆楠, 王晓慧等. 线粒体脑肌病伴乳酸血症和卒中样发作综合征的临床特征及遗传学研究. 中国循证儿科杂志. 2008.(03): 169-176.
[17]Iizuka T, Sakai F, Suzuki N, et al. Neuronal hyperexcitability instroke-like episodes of MELAS syndrome. Neurology. 2002. 59(6):816-24.
[18]Thambisetty M, Newman NJ. Diagnosis and management of MELAS. ExpertRev Mol Diagn. 2004. 4(5): 631-44.
[19]Anan R, Nakagawa M, Miyata M, et al. Cardiac involvement inmitochondrial diseases. A study on 17 patients with documentedmitochondrial DNA defects. Circulation. 1995. 91(4): 955-61.
[20]焉传祝. 线粒体病诊断中的若干问题. 中华神经科杂志. 2005. (08): 533-534.
[21]郭玉璞. 线粒体脑肌病. 中华神经科杂志. 1997. (05): 46-50.
[22]Rahman S, Poulton J, Marchington D, Suomalainen A. Decrease of 3243A-->G mtDNA mutation from blood in MELAS syndrome: alongitudinal study. Am J Hum Genet. 2001. 68(1): 238-40.
[23]Tokunaga M, Mita S, Murakami T, et al. Single muscle fiber analysisof mitochondrial myopathy, encephalopathy, lactic acidosis, andstroke-like episodes (MELAS). Ann Neurol. 1994. 35(4): 413-9.
[24]陈合成, 杨金升, 石向群, 王娟, 杨银升. 线粒体脑肌病的临床、病理与神经电生理. 卒中与神经疾病. 2007. (01):28-31.
[25]戚晓昆, 郭玉璞, 王湘庆. MELAS型线粒体脑肌病的临床、病理及影像学研究. 中华神经科杂志. 2001. (04):39-41+71.
[26]Fujii T, Okuno T, Ito M, et al. CT, MRI, and autopsy findings inbrain of a patient with MELAS. Pediatr Neurol. 1990. 6(4):253-6.
[27]Tokunaga M, Mita S, Sakuta R, Nonaka I, Araki S. Increasedmitochondrial DNA in blood vessels and ragged-red fibers inmitochondrial myopathy, encephalopathy, lactic acidosis, andstroke-like episodes (MELAS). Ann Neurol. 1993. 33(3): 275-80.
[28]Goto Y, Horai S, Matsuoka T, et al. Mitochondrial myopathy,encephalopathy, lactic acidosis, and stroke-like episodes (MELAS):a correlative study of the clinical features and mitochondrial DNAmutation. Neurology. 1992. 42(3 Pt 1): 545-50.
[29]吴磊, 吴卫平, 黄德晖, 徐全刚, 蒲传强. 误诊为单纯疱疹病毒性脑炎的MELAS综合征8例临床分析. 临床神经病学杂志.2006. (06): 417-419.
[30]Apostolova LG, White M, Moore SA, Davis PH. Deep white matterpathologic features in watershed regions: a novel pattern ofcentral nervous system involvement in MELAS. Arch Neurol. 2005.62(7): 1154-6.
[31]Scaglia F, Wong LJ, Vladutiu GD, Hunter JV. Predominant cerebellarvolume loss as a neuroradiologic feature of pediatric respiratorychain defects. AJNR Am J Neuroradiol. 2005. 26(7): 1675-80.
[32]Kim IO, Kim JH, Kim WS, Hwang YS, Yeon KM, Han MC. Mitochondrialmyopathy-encephalopathy-lactic acidosis-and strokelike episodes(MELAS) syndrome: CT and MR findings in seven children. AJR Am JRoentgenol. 1996. 166(3): 641-5.
[33]Mizrachi IB, Gomez-Hassan D, Blaivas M, Trobe JD. Pitfalls in thediagnosis of mitochondrial encephalopathy with lactic acidosis andstroke-like episodes. J Neuroophthalmol. 2006. 26(1): 38-43.
[34]Iizuka T, Sakai F. Pathogenesis of stroke-like episodes in MELAS:analysis of neurovascular cellular mechanisms. Curr Neurovasc Res.2005. 2(1): 29-45.
[35]Moller HE, Kurlemann G, Putzler M, Wiedermann D, Hilbich T, FiedlerB. Magnetic resonance spectroscopy in patients with MELAS. J NeurolSci. 2005. 229-230: 131-9.
[36]Abe K, Yoshimura H, Tanaka H, Fujita N, Hikita T, Sakoda S.Comparison of conventional and diffusion-weighted MRI and proton MRspectroscopy in patients with mitochondrial encephalomyopathy,lactic acidosis, and stroke-like events. Neuroradiology. 2004.46(2): 113-7.
[37]Lin DD, Crawford TO, Barker PB. Proton MR spectroscopy in thediagnostic evaluation of suspected mitochondrial disease. AJNR Am JNeuroradiol. 2003. 24(1): 33-41.
[38]Cross JH, Gadian DG, Connelly A, Leonard JV. Proton magneticresonance spectroscopy studies in lactic acidosis and mitochondrialdisorders. J Inherit Metab Dis. 1993. 16(4): 800-11.
[39]Iizuka T, Sakai F, Kan S, Suzuki N. Slowly progressive spread ofthe stroke-like lesions in MELAS. Neurology. 2003. 61(9):1238-44.
[40]Nariai T, Ohno K, Akimoto H, et al. Cerebral blood flow, vascularresponse and metabolism in patients with MELAS syndrome--xenon CTand PET study. Keio J Med. 2000. 49 Suppl 1: A68-70.
[41]Finsterer J. Treatment of central nervous system manifestations inmitochondrial disorders. Eur J Neurol. 2010 .
[42]Marriage B, Clandinin MT, Glerum DM. Nutritional cofactor treatmentin mitochondrial disorders. J Am Diet Assoc. 2003. 103(8):1029-38.
[43]Chinnery P, Majamaa K, Turnbull D, Thorburn D. Treatment formitochondrial disorders. Cochrane Database Syst Rev. 2006. (1):CD004426.
[44]Barisic N, Bernert G, Ipsiroglu O, et al. Effects of oral creatinesupplementation in a patient with MELAS phenotype and associatednephropathy. Neuropediatrics. 2002. 33(3): 157-61.
[45]De Stefano N, Matthews PM, Ford B, Genge A, Karpati G, Arnold DL.Short-term dichloroacetate treatment improves indices of cerebralmetabolism in patients with mitochondrial disorders. Neurology.1995. 45(6): 1193-8.
[46]熊杰, 徐平湘, 孙丽娜, 成亮, 冯亦璞, 王晓良. 丁基苯酞对原代培养神经元线粒体功能的保护作用. 中药药理与临床. 2007.(05): 73-76.
[47]Cejudo P, Bautista J, Montemayor T, et al. Exercise training inmitochondrial myopathy: a randomized controlled trial. MuscleNerve. 2005. 32(3): 342-50.
[48]Taivassalo T, Haller RG. Exercise and training in mitochondrialmyopathies. Med Sci Sports Exerc. 2005. 37(12): 2094-101.
[49]Doyle SR, Chan CK. Mitochondrial gene therapy: an evaluation ofstrategies for the treatment of mitochondrial DNA disorders. HumGene Ther. 2008. 19(12): 1335-48.
该文发表在《临床神经病学杂志》上,请勿转载抄袭!