糖原累积病
来自 维客
Jump to: navigation, search
糖原累积病
glycogen storage diseases
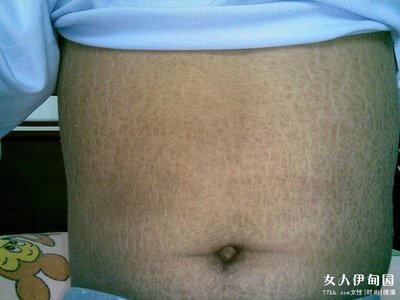
一组少见的遗传性糖原代谢紊乱的疾病。又称糖原沉着症、糖原累积病。因糖原分解与合成有关的某些酶系统缺乏,糖原分解困难,糖原异常地沉积于全身各组织,尤其是肝脏、心脏及肌肉中。表现为肝脏肿大,或伴有低血糖和高血脂、血清乳酸增高、心脏扩大、肌张力降低、肾肿大、高尿酸血症及肌红蛋白尿,中枢神经症状包括运动障碍、智力差。确诊需依靠酶的测定。本病为常染色体隐性遗传。常见于男性,多在婴儿期发病,儿童期死亡,少数患者可活到成年。目前尚缺乏有效治疗,主要是对症处理。
糖原代谢人体内的碳水化合物以糖原形式储存。糖原是分子量高达300~1000万、由1万个以上葡萄糖单位聚合而成的高分子多糖,有许多分支,而呈树枝状(见图),直链部分以α-1,4-糖苷键相连,侧链处又以α-1,6-糖苷键连接。糖原分子中心有葡萄糖残基还原端,在分枝外周有许多非还原端,糖原的合成和分解就在非还原端进行。糖原分解需要磷酸化酶催化,最先是非还原端α-1,4-糖苷键的糖苷基端磷酸化,水解为1-磷酸葡萄糖,1-磷酸葡萄糖在磷酸葡萄糖变位酶催化下变为6-磷酸葡萄糖(G-6-P),然后G-6-P在6-磷酸葡萄糖酶影响下变为葡萄糖。糖原分解的另一途径由细胞溶酶体中的α-1,4-糖苷酶来完成。糖原生成从葡萄糖开始,首先葡萄糖由己糖激酶催化为6-磷酸葡萄糖,再经过磷酸葡萄糖变位酶催化,生成1-磷酸葡萄糖,1-磷酸葡萄糖在二磷酸尿嘧啶核苷(UDP)、葡萄糖焦磷酸化酶影响下生成UDP-葡萄糖,然后经过糖原合成酶及分支酶协同催化下,最后形成糖原。组织(肝、骨骼肌、心肌、平滑肌)中的糖原等细胞内外液的葡萄糖经常保持动态平衡,保证葡萄糖不间断地适当供应全身各组织器官,特别对脑组织是一刻不能缺少的。
临床分型 依据缺陷的酶不同可分为10型及酶正常的Ⅺ型,又可根据糖原累积的主要器官分为肝型、心型、肌型。其中以肝型较多见,肝型包括Ⅰ型、Ⅲ型、Ⅳ型、Ⅵ型、Ⅷ型;心型为Ⅱ型;肌型为Ⅴ型及Ⅶ型。症状的轻重取决于糖原转化为血中葡萄糖的障碍程度。疾病的最后确诊依靠酶测定。Ⅱ型、Ⅲ型、Ⅳ型可通过羊水穿刺测酶及羊水细胞培养作产前诊断。
①糖原累积病Ⅰ型,又称肝型糖原累积病。1929年首先由E.O.K.冯•吉尔克描述,故又名冯•吉尔克氏病。常累及肝、肾。1952年发现本病患者肝内缺乏6-磷酸葡萄糖酶,造成糖原分解障碍,肝脏不能从糖原、乳酸、氨基酸形成葡萄糖,引起空腹血糖低,而低血糖抑制胰岛素分泌,脂蛋白分解减少,脂蛋白酶活性降低,使脂肪大量动员,刺激脂肪分解,形成高甘油三酯血症。由于糖酵解增多,形成乳酸增多。脂肪酸在肝内氧化不全,血中酮体增多,可出现酮症酸中毒。由于慢性酸中毒引起负钙平衡,可发生骨质疏松。又因己糖旁路代谢增强,尿酸合成增加,引起高尿酸血症及痛风。本型主要侵犯婴儿、儿童、成人,症状随年龄有所不同,两性均可受累。
诊断除根据临床表现外,可作胰岛高血糖素耐量试验,肌注肾上腺素后血糖不升高,而乳酸增高,这有助于诊断。确诊需作肝组织活检,可见肝细胞肿大,糖原增加常超过5%,6-磷酸葡萄糖酶活性降低或消失。
饮食宜少量多餐饮,以避免低血糖和防止酸中毒。膳食中蛋白质含量正常,脂肪要低,总热量不宜过高,以防止肥胖。血清乳酸高的病人宜服碳酸氢钠,以防止酸中毒。有的病人可作门腔静脉吻合术,以将血糖转移至全身,可较好利用,这对纠正代谢紊乱与生长发育也有裨益。
②糖原累积病Ⅱ型,又称蓬普氏病。是最严重的一型。1963年发现。病因为溶酶体中α-1,4-葡萄糖苷酶(即酸性麦芽糖酶)缺陷。α-1,4-葡萄糖苷酶存在于各组织细胞溶酶体内,缺乏此酶即不能分解糖原,以致糖原累积在溶酶体内,实际上是容酶体累积病。糖原大量累积于心肌、骨骼肌等全身组织,引起心脏增大,心脏重量可达正常的2~5倍,故又称心型糖原累积病。患儿出生后半年之内出现紫绀、呼吸困难,呛咳常见,常因心力衰竭或支气管肺炎于1岁内死亡。成年患者症状轻微,表现为慢性全身肌无力,肌张力低下。诊断有赖于肝脏和肌肉活检,电子显微镜检查显示糖原颗粒沉积增多,肝糖原颗粒外有一容酶体膜存在。肝、肌肉及白细胞内α-1,4-葡萄糖苷酶均缺乏,皮肤活检后培养的纤维细胞中亦缺乏此酶,据此可确诊。目前尚缺乏有效治疗,有人研究用白细胞提取酶替 代治疗。
③糖原累积病Ⅲ型,又称科里氏病。由于淀粉1,6-糖苷酶即脱支酶缺乏,而1-磷酸葡萄糖酶和磷酸化酶均正常,因此糖原分子只能分解到第一层分支为止,不能完全分解,结果在许多组织中聚集结构异常的糖原。过多的糖原累积于肝、心肌和其他肌肉组织。本型比I型症状要轻,但较常见。多见于幼儿,表现肝脏肿大,发育障碍,至青春期肝脏可恢复正常大小。血脂升高,但尿酸正常。成人症状较轻,仅有肌肉无力或无症状。由于糖原异生正常,故空腹低血糖少见。根据白细胞、红细胞、肝脏、肌肉及心肌中淀粉1,6-糖苷酶缺乏即可确诊。肾上腺素及胰高血糖素试验接近正常。本型预后良好,主要对症治疗。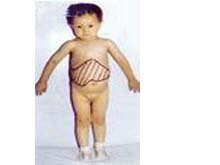
④糖原累积病Ⅳ型,又称安德森氏病。罕见,系淀粉1,4→1,6-转糖酶即分支酶缺乏,所累积的糖原结构异常,分支减少。临床表现为进行性肝硬变、腹水、空腹血糖低、肝功能异常。病儿2岁左右夭折。若新生儿有肝硬变即应疑及本病。预后差,无特效治疗。
⑤糖原累积病Ⅴ型,又称肌型糖原累积病、麦卡德尔氏病。因肌磷酸化酶缺乏,肌糖原分解困难,糖原累积,有关糖代谢的其他酶系统正常。患者以男性占多数,家族中兄弟姐妹可患同样疾病,患者往往有近亲结婚史。一般儿童期无症状,待至10余岁发病。由于酶缺乏,正常合成的糖原不能在肌肉内作为燃料,因此剧烈运动后出现肌肉酸痛、四肢僵硬、肌肉痉挛,有时引起肌红蛋白尿,甚至肾功能衰竭。可作运动前后缺血性乳酸试验,即以血压绷带维持血压于收缩压,同时使手指伸张握拳,比较运动前后血乳酸浓度,正常人运动后血乳酸浓度增高,而本病患者不升高,原因为缺乏磷酸化酶,肌糖原未能分解。肌肉活检发现有肌糖原累积及肌磷酸化酶缺乏即可确诊。无特效治疗,平时避免剧烈运动,可防止发作。
⑥糖原累积病Ⅵ型,又称赫尔氏病。因肝缺乏磷酸化酶,不能分解肝糖原产生葡萄糖,维持血糖水平,并使肝糖原累积于肝。肌磷酸化酶正常。临床表现以肝肿大为特征,低血糖症状一般较轻或缺如。诊断除依靠肝组织活检及肝磷酸化酶分析外,白细胞磷酸化酶鉴定也有助于诊断。本型病情轻,预后好,可不予治疗。
⑦糖原累积病Ⅶ型,又称垂井氏病。由于缺乏肌肉磷酸果糖激酶,糖原合成障碍,肝糖原太少。肌肉磷酸果糖激酶活性为正常的1~3%。症状似V型。重体力劳动后发生肌痉挛和肌疼痛。由于约50%红细胞中的酶活性消失,红细胞寿命缩短,有网织红细胞增高。确诊主要靠肌肉活组织检查或查血红细胞,鉴定此酶缺乏与否。本型预后较好,但无特效治疗。
⑧糖原累积病Ⅷ型,肝磷酸化酶激酶缺乏所致。为性联遗传,也有学者认为因肝磷酸化酶激酶呈失活状态。磷酸化酶激酶可催化磷酸化酶b(无活性),使之转变成磷酸化酶a(活性型)。正常肝内60%磷酸化酶有活性,缺乏此酶,磷酸化酶b转化为磷酸化酶a的数量降低,糖原分解发生障碍,肝糖原累积于肝和肌肉。表现无症状的肝肿大、肢体僵硬、去大脑状态、肌无力、血清转氨酶升高、血脂升高、低血糖,空腹血乳酸可正常,摄入碳水化合物后血乳酸升高,胰高血糖素试验显示正常反应。在白细胞、红细胞及肝脏中缺乏磷酸化酶激酶b,即可确诊。患者大多数为男性,一般至婴幼儿期夭折。无特殊治疗。
⑨糖原累积病Ⅸ型。少见。肝磷酸化酶激酶缺陷,常累及肝脏,表现肝大,无低血糖酸中毒。为常染色体隐性遗传。
⑩糖原累积病 X型。少见。依赖cAMP的磷酸化酶激酶缺陷,常累及肝、肌肉,表现肝大,肝、肌肉糖原沉积。
Ⅺ型。所有酶的活性均正常。常累及肝或肾,表现肝大,伴有范可尼氏综合征、维生素D 抵抗性佝偻病、遗传性果糖不耐受症等。
________________________________________
糖原累积病 glycogen storage disease
一组少见的遗传性糖原代谢紊乱的疾病。又称糖原沉着症、糖原累积病。因糖原分解与合成有关的某些酶系统缺乏,糖原分解困难,糖原异常地沉积于全身各组织,尤其是肝脏、心脏和肌肉中。表现为肝脏肿大,可伴有低血糖、高血脂、血清乳酸增高、心脏扩大、肌张力降低、肾肿大、高尿酸血症及肌红蛋白尿,中枢神经症状包括运动障碍、智力差。确诊需依靠酶的测定。本病为常染色体隐性遗传。常见于男性,多在婴儿期发病,儿童期死亡,少数可活到成年。尚无特效疗法,主要是对症处理。
取自"http://www.wiki.cn/wiki/%E7%B3%96%E5%8E%9F%E8%B4%AE%E7%A7%AF%E7%97%85"