常用的分子生物学基本技术
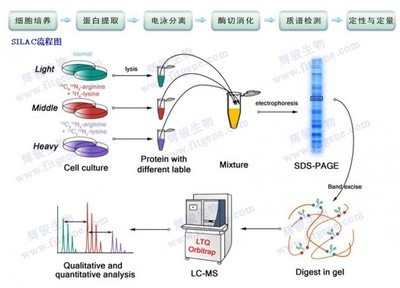
核酸分子杂交技术
由于核酸分子杂交的高度特异性及检测方法的灵敏性,它已成为分子生物学中最常用的基本技术,被广泛应用于基因克隆的筛选,酶切图谱的制作,基因序列的定量和定性分析及基因突变的检测等。其基本原理是具有一定同源性的原条核酸单链在一定的条件下(适宜的温室度及离子强度等)可按碱基互补原成双链。杂交的双方是待测核酸序列及探针(probe),待测核酸序列可以是克隆的基因征段,也可以是未克隆化的基因组DNA和细胞总RNA。核酸探针是指用放射性核素、生物素或其他活性物质标记的,能与特定的核酸序列发生特异性互补的已知DNA或RNA片段。根据其来源和性质可分为cDNA探针、基因组探针、寡核苷酸探针、RNA探针等。
固相杂交
固相杂交(solid-phasehybridization)是将变性的DNA固定于固体基质(硝酸纤维素膜或尼龙滤膜)上,再与探针进行杂交,故也称为膜上印迹杂交。
斑步杂交(dot hybridization)
是道先将被测的DNA或RNA变性后固定在滤膜上然后加入过量的标记好的DNA或RNA探针进行杂交。该法的特点是操作简单,事先不用限制性内切酶消化或凝胶电永分离核酸样品,可在同一张膜上同时进行多个样品的检测;根据斑点杂并的结果,可以推算出杂交阳性的拷贝数。该法的缺点是不能鉴定所测基因的相对分子质量,而且特异性较差,有一定比例的假阳性。
印迹杂交(blotting hybridization)
Southern印迹杂交:凝胶电离经限制性内切酶消化的DNA片段,将凝胶上的DNA变性并在原位将单链DNA片段转移至硝基纤维素膜或其他固相支持物上,经干烤固定,再与相对应结构的已标记的探针进行那时交反应,用放射性自显影或酶反应显色,检测特定大小分子的含量。可进行克隆基因的酶切图谱分析、基因组基因的定性及定量分析、基因突变分析及限制性长度多态性分析(RELP)等。
Northern印迹杂交:由Southerm印杂交法演变而来,其被测样品是RNA。经甲醛或聚乙二醛变性及电泳分离后,转移到固相支持物上,进行杂交反应,以鉴定基中特定mRNA分子的量与大小。该法是研究基因表达常用的方法,可推臬出癌基因的表达程度。
差异杂交(differential hybridization)
是将基因组文库的重组噬菌体DNA转移至硝酸纤维素膜上,用两种混合的不同cDNA探针(如:转移性和非转移性癌组织的mRNA逆转录后的cDNA)分别与滤膜上的DNA杂交,分析两张滤膜上对应位置杂交信息以分离差异表达的基因。适用于基因组不太复杂的真核生物(如酵母)表达基因的比较,假阳性率较低。但对基因组非常复杂的盐酸核生物(如人),则因工作量太大,表达的序列所占百分比较低(仅5%左右),价值不大。
cDNA微点隈杂交(cDNA microarray hybridization)
是指将cDNA克隆或cDNA的PCR产物以高度的列阵形式排布并结合于固相支持物上(如:尼龙膜或活化的载玻片)以微点阵,然后用混合的不同DNA探针与微点阵上的DNA进行杂交。再利用荧光、化学发光、共聚焦显微镜等技术扫描微点阵上的杂交信息。它比差异杂交技术的效率高、速度快、成本低,适用于大规模的分析。已成商品问世。其缺点是无法克服保守的同源序列及重序对杂交信息的干扰。
寡核苷酸微点隈杂交(oligonucleotide microarray hybridization)
是在特殊的固相支持物上原位合成寡核苷酸,使它共价结合于支持物表面,与平均长度为20-50nt的混合RNA或cDNA探针进行杂交,以提高杂交的特异性和灵敏度。应用共聚焦显微镜可检测跨越三个数量级的杂交信息。适用于低丰度mRNA的检测,以区分基因家族不同成员的差异表达特征,或鉴定同一转录在不同组织和细胞中的选择性剪接。但有工作量较大、成本较高、速度较慢等弱点。
液相杂交(solution hbridization)
指使变性的待测核酸单链与放射笥核素标记的已知的酸单链(探针)在溶液中保温,使之形成杂交复合物。反应结束后,用羟磷灰石法或酶解法将未被杂交的单链和杂交双链分开,然后结膈后在杂妆分子中的探针量,就可推算出被测的核酸量。
递减杂交(subtractive hybridizatio)
是利用两种来源一致而功能不同的组织细胞提取mRNA(或逆转录成的cDNA),在一定的条件下以过量的驱动mRNA或cDNA与测试的单链cDNA或mRNA进行液相杂交,通过羟基磷灰石柱层析筛选除去两者间同源的杂交体。经多轮杂交策选、除去两者之间相同的基因成分,保留特异表达的目的基因或工基因片段。以后者筛选cDNA文库,可获得特异表达的目的基因cDNA全长序列。
核酸原位杂交
用特定标记的已知顺序核酸作为探针与细胞级或组织切片中核酸进行复性杂交并对其实行检测的方法,称为核酸原位杂交(nucleic acidhybridization insitu)。用来检测DNA在细胞核或染色休上的分布,与细胞内RNA进行杂交以研究该组织细胞中特定基因表达水闰;还用于细胞、组织中有无特异性菌、病毒检测的研究。
该法的优点是特异性高,可精确定位;能在成分复杂的组织中进行单一细胞的研究而不受同一组织中其他成分的影响;不需要从组织中提取核酸,对于组织中含量极低的靶序列有极高的敏感性;并可完整地保持组织与细胞的形态。因此被广泛应用于医学生物学的研究,如基因定位、基因制失,基因易位、特异基因整合部物色检测等研究。近年来由定性发展到定量,方法更为完善。
DNA分子克隆技术(也称基因克隆技术)
在体外将DNA分子片段与载体DNA片段连接,转入细胞获得大量拷贝的过程中DNA分子克隆(或基因克隆)。其基本步骤包括:制备目的基因→将目的基因与载体用限制性内切酶切割和连接,制成DNA重组→导入宿主细胞→筛选、鉴定→扩增和表达。载体(vecors)在细胞内自我复制,并带动重组的分子片段共同增殖,从而产生大量的DNA分子片段。主要目的是获得某一基因或NDA片段的大量拷贝,有了这些与亲本分子完全相同的分子克隆,就可以深入分析基因的结构与功能,随着引入的DNA片段不同,有两种DNA库,一种是基因组文库(genomiclibrary),另一种是cDNA库。
载体 所谓载体是指携带靶DNA片段进入宿主细胞进行扩增和表达的工具。
细菌质粒是一种细菌染色体外小型双链环状结构的DNA,分子大小为1-20kb,对细菌的某些代谢活动和抗药性表型具有一定的作用。质粒载体是在天然质粒的基础上人工改造拼接而成。最常用的质粒是pBR322。
噬菌体(phaeg)噬菌体是感染细菌的病毒,按其生活周期分为溶菌型及溶原型两型。用野生型λ噬菌体改造和构建的噬菌体载体是线性双链DNA,基因组约为50kb,最常用的哓菌体为λDNA及其衍生系列。
黏性质粒(cosmid) 是由质粒与噬菌体λDNA组成的一种4-6kb的环状杂种DNA。
表达型载体 将上述细菌质粒载体或噬菌体载体接上启动基因及多聚核糖体的基因序列组成了表达型载体。
基因库的建造
含有某种生物体全部基历的随机片段的重组DNA克隆群体,其含有感光趣的基因片段的重组子,可以通过标记探针与基因库中的重组子杂交等方法而筛选出来,所得到的克隆经过纯化和扩增,可用于进一步的研。其主步骤包括:(1)构建基因库迅速的载体;(2)DNA片段的制备;(3)DNA片段与载体DNA的连接;(4)包装和接种。
cDNA库的建造
是指克隆的DNA片段,是由逆转录酶自mRNA制备的cDNA。cDNA库包括某特定细胞的全部cDNA克隆的文库,不含内含子。
特异基因的筛选
常用的方法有:(1)克隆筛选即探针筛选法;(2)抗体检测法,检测其分泌蛋白质来筛选目的基因;(3)放射免疫筛选法,查出分泌特异抗原的基因;(4)免疫沉淀法,进行特异基因的筛选。
核酸序列测定
DNA的碱基序列决定着基因的特性,DNA序列分析(测序,sequencing)是分子生物学重要的基本技术。无论从基因库中筛选的癌基因或经PCR法扩增的基因,最终均需进行核酸序列分析,可藉以了解基因的精细结构,获得其限制性内切酶图谱,分析基因的突变及对功能的影响,帮助人工俣成基因、设计引物,以及研究肿瘤的分子发病机制等。测序是在高分辨率变性聚丙烯酰胺凝胶电泳技术的基础上建立起来的。目前最常用的方法有Maxam-Gilbert的化学降解少和Sanger的双脱氧法等,近年来已有DNA序列自动测定仪问世。化学降解法是在DNA的片段的5`端标记核素,然后用专一性化学试剂将DNA特异地降解,在电泳和自显影后,可得到从标记端延伸的片段供测读序列和进行比较。一般能读出200-250个核苷酸序列。双脱氧法是采用核苷酸链终止剂,如:2`,3`-双脱氧核苷三磷酸ddNTP(如ddTTP、ddTTP、ddGTP和ddCTP中的一种)掺入到DNA链中以终止链的延长,与掺入4种正常的dNTP的混合物分成四组进行反应,这样可得到一组结尾长衙不一、不同专一性核苷酸链终止剂结尾的DNA片段,经凝胶电泳分离和放射自显影,可读出合成的DNA核苷酸序列,根据碱基互补原则,可推算出模板DNA分子的序列。
化学降解法只需一化学试剂,重复性好,容易掌握;而双脱氧法需单链模板、特异的寡核苷酸引物及高质量的DNA聚合酶,便随着M13噬菌体载体的发明和运用,合成的引物容易获得,测序技术不断改进,故此法已被广泛应用。基脱氧法的自动激光荧光测序仪,使测工作更快速和简便,而且保证高度重复性。至于RNA测序现大多采用将mRNA逆转录成cDNA后同测序,然后反推RNA序列
聚合酶链反应技术
聚合酶链反应技术简称PCR技术,是一种利用DNA变性和复性原理在体外进行特定的DNA片断高效扩增的技术,可检出微量靶序列(甚至少到1个拷贝)。在模板DNA、引物和四种脱氧核糖核苷酸存在的条件下依赖于DNA聚合酶的酶促合成反应。仅需用极少量模板,在一对引物介导下,在数小时内可扩增至100万-200万份拷贝。PCR反应分三步:变性、退火及延伸。以上三步为一循环,每一循环的产物作为下一个的模板,这样经过九小时的循环,每一循环的产物作为下一个循环的模板,这样经过数上时的循环后,可得到大量复制的特异性DNA片段。反应条件一般为94℃变性30秒,55℃退火30秒,70-72℃延伸30-60秒,共进行30次循环左右,最后再延伸72℃5分钟,4℃冷却终止反应。
1.逆转录PCR(RT-PCR)用来扩增RNA的方法。
2.竞争逆转录PCR(competitive reverse transcription-polymerase chainrection C-RT-PCR)低丰度RNA定量的好方法。
3.多重PCR(multiplesPCR)在同一PCR体系中加入多对引物可用于基天长度很长,发生多处缺失的觳狻@┰鐾荒0宓募父銮颉?br>
4.多种PCR可同时加入多套以生物素标记的引物起进手PCR反应。
5.反向PCR(iverse polymerase chainreaction)对一个已知的DNA片段两侧的未知序列进行扩增和研究。
6.不对称PCR在扩增循环中引入不同引物浓度,以得到单链DNA并进行列测定,以了解目的基因的序列。
7.锚定PCR(anchored polymerase chainreaction)帮助克服序列未知或序列未全知带来的障碍。在未知序列未端添加同聚物尾序,将互补的引物连接于一段带限制性内切酶位点的锚上,在锚引物和基因另一侧特异性引物的作用下,将未知序列扩增出来。
8.着色互补试验或荧光PCR(color complementation assay or fluorescentPCR)原理是用不同荧光染粒,分别标记于不同寡核苷酸引物上,同时扩增多个DNA片段,反应完毕后,利用分子筛选去多余的引物。用紫外线照射扩增产物,就能显示某一DNA区带荧光染料颜色的组合,如果某一DNA区带荧光染色料颜色的组合,如果某一DNA区带缺失,则会缺乏相应的颜色。
9.双温PCR(two-temperaturePCR)仅仅执行两步温度程序。合并退火与延伸温度。一般常用温度是94-95℃和46-47℃。可以提高反应的速度和特异性。
上述这些PCR技术的应用:(1)PCR技术扩增特定序列为基础,采用多种方法进行检测,如PCR-RFLP、PCR-SSCP从已克隆的双链DNA中产生特异性序列作为分子探针;(2)通过选择性扩增cDNA中产生特异性序列作为分子探针;(2)通过选择性扩增cDNA中的特殊片段,产生针对尚未克隆的基因的探针;(3)从微量mRNA中产生cDNA文库;(4)产生大量用于序列分析的DNA;(5)分析基因突变;(6)做染色体步移。
10.原位PCR技术:PCR虽然能扩增包括福尔马林固定、石蜡包埋组织的各种标本的DNA,但扩增的DNA或RNA产物不能在组织细胞中定位,因而不能直接与特定的组织细胞特征相联系,这是该技术一个局限性。原位杂交虽具有良好的定位能力,但由于其敏感性问题,尤其是在石蜡切片中,尚不能检测出低含量的DNA或RNA序列。而原位PCR(Insitu PCR,简称ISPCR),它可使扩增的特定DNA片段在分离细胞和组织切片中定位,从而弥补了PCR和原位杂交的不足,具有良好的应用前景。目前,已有多种设计的原位PCR扩增仪系统问世,使操作简便,软件灵活,已成为拉增固定细胞和石蜡包埋组织中特定DNA和RNA序列的有用工具。
IS PCR的技术特点
(1)既具有PCR的特异性与高灵敏性,又具有原位杂交的定位准确性;(2)测到低于2个拷贝量的细胞内特定DNA序列,甚至可检测出单一细胞中的仅含一个拷贝的原病毒DNA;(3)有助于细胞内特定核酸序列定位与其形态学变化的结合分析;(4)可用于正常或恶性细胞,感染或非感染细胞的鉴定与区别。
IS PCR的基本方法
IS-PCR是PCR和原位杂交两种技术的绫事,实质是一种将靶DNA或RNA在原位扩增后再进行原位杂交的技术,操作程序基本兼有两种技术的所有过程,首先在适当处理的细胞和组织切片上滴加PCR反应液,然后把载玻片放入热循环仪中进行扩增,最后通过标记的探针进行原位杂交检测扩增产物。
初步应用
有关原位PCR技术应用成功的第一篇报道是对感染绵羊中枢神经系统visna病毒在绵羊脉络从一细胞中检查,通过PCR扩增,仅用150碱基对的短探针就可通过ISH检查到了细胞内的visna病毒。从开始在试管内细胞行PCR扩增后再涂片做ISH(原位杂交)检查,已发展到用福尔马林固定、石蜡切片的常规病理组织方法做原位PCR检查。有传统的标记探针的原位PCR法(间接法),也有将标记物先标记PCR的引物,让标记物扩增后再行ISH检查的直接法。因此,目前就原位PCR方法而言,尚未能获得统一,也即未能比较出哪一种方法最可靠简便,各家报道的原位PCR技术中,在标本处理和种类上尚不同(细胞悬液、涂片、组织切片等),想检测的靶核酸也不同(病毒或体细胞的DNA或mRNA),扩增的方法上有不同(单引物、多引物),最后显示的方法也不同(直接法、间接法)。这些还都有待在今后的工作中积累经验,以求一致。
原位PCR应用的课题,目前正片于开始阶段,还很局限,对人体B细胞淋巴瘤中Ig单拷贝基因重组的检查以及对人体外周血中单核细胞HLA-DQ不同亚型的测定,归纳起来,主要应用于两方面;(1)检测外源怀基因片段,集中在病毒感染的检查上,如HIV、HPV、HBV、CMV等;(2)内源性基因片段,如人体的单基因病、重组基因、易位的染色体、Ig的mRNA片段、癌基因片段等。
基因转染技术
将特定的遗传信息传递到真核细胞中,这种能力不但革新了生物学和医学中许多基本问题的研究,也推动了诊断和治疗方面的分子技术发展,并使基因治疗成为可能。目前基因转移技术已广泛用于基因的结构和功能分析、基因表达与调控、基因治疗与转基因动物等研究。本部分将介绍目前基因转移的主要方法;重点介绍基因转移的病毒方法的基因转染(transfection)技术。
基因运载系统
将某一特定的靶基因传递到靶细胞,需要应用某一基因运载系统,目前将这一系统概括为两大类:非病毒辣方法和病毒方法。
1.基因转移的非病毒方法
直接注射法 将含有DNA的溶液直接注射到肌肉,以引起邻近的细胞摄入DNA燕进行表达,在肌细胞中,基因表达可持续数月。
磷酸钙共沉淀法 将氯化钙、DNA和磷酸盐缓冲液混合,形成磷酸钙微沉淀,附着于细胞膜并经过细胞内吞作用耐是入细胞质。该方法的转化效率通常很低。
脂质体染法 指质体能在体内或体外提供运载外源性遗传物质进入细胞的载体。脂质体介导的基因转移的最大优势在于能在活体内应用。
受体介导的基因转移依靠受体介导的细胞内吞途径以转移外源基因。受体介导的基因转移方法是在质粒DNA和某种特异的多肽(配体)之间形成复合体,而这种多肽能为细胞表面的肥体所识别。如若将DNA在体内运送至肝内,可以选将DNA和能与肝细胞受体特异结合的去唾液酸糖蛋白质偶联,以便通过细胞内吞过程而被摄入,这种DNA大部分被肝脏所摄取。应用该方法转移的外源基因在活体内的表达持续时间较短,在评估实际应用前影上还在一些问题。
显微注射法在显微镜下,将DNA经同细胞玻璃针地接注入细胞,该法适合于各种培养生长的细胞,但需要一定的设备和操作用支巧。
电穿孔法 利用脉冲场将DNA导入培养细胞。
微粒子轰击法(基因枪)近几年发展较快的转移方法。使用高能微粒子轰击将DNA导入培胞或活的哺乳动物组织内。亚微粒的钨和金能自发地吸附DNA,将这些微弹加强速到很的速度轰击细胞。实验结果表明,用这种方法靶基因可在皮肤、肌肉、肝、胰、胃和乳腺等细胞中表达。
胚胎干细胞法胚胎干细胞是从受精卵开始分裂至4个国胞阶段未分化和具有多种潜能的生殖细胞,能在体外培养,可作为正面细胞,制备转基因动物,以研究基因定向整合或基因剔险及基因及能。
精子载体法最近发现用精子和NDA-剂孵育,可捕获得DNA。通过受精过程,将外源性基因导入受精卵,可捕获得物物,大大间化了转基因动物的制备过程。
2.基因转移的病毒的方法
病毒作为基因转移的是基因递送有有并工具,病毒载体的优点有:(1)在转化的细胞中传播重组的DNA分子作为稳定的遗传成分;(2)在可能将有有制陷的或突变的基因置于病毒调节信号的控制下以进行研究;(3)能将克隆的基因作为病毒微染色体的一部分,并能进分离;(4)转移效率较高。其主要缺点是病毒载体对外源基因的最大容纳量只有2500bp。目前常用的病毒载体有下列几种。
逆转录病毒载体逆转录病毒辣为RNA病毒,它们的基因组编码在一条单链RNA他妇上,病毒进入细胞通过逆转录作用,病毒RNA即转变为双链DNA分子,DNA进入细胞核并整合在细胞染色体中,这种整合的病毒称为原病毒。在原病毒的两端各有一长末端重复序列(LTR),LTR内侧还有为复制所必需的其他顺序,包括包装信号。目前常用的逆转病毒载体有CMV和SV。
DNA病毒载体主要有腺病毒相关病毒载体,腺端正毒载体,疱疹病毒载体。对DNA病毒载体的研究是当前基因基因治疗研究中的重点领域。
常用的基因转染技术
将外源基因导入靶细胞需要一定的载体和导入方法,基因转技术则是将纯化的含有靶基因的质粒DNA送入细胞内,并在细胞内表达。转染方法有多种,根据不同的细胞,贴壁或悬浮细所可选用不同的方法,其目的是要达到设置转染效率,影响转染产率的因素有多种,包括转染方法、操作技术、质粒DNA的纯度、靶细胞的生长状态等,下面重点介绍向几种常用的转染技术:
靶细胞的准备被用于作靶基因转染的细胞,其生长状态如何,将直接决定了基因转染效率。如为贴壁生长的细胞,一般要求在转染前一日,必需应用胰酶处理成单细胞悬液,重新接种于培养皿或瓶,细胞密度以铺满培养器皿的60%为宜,转染当日,在转染前4小时换一将近新鲜培养液。对于悬浮细胞,也需在转染前4小时换一次新鲜培养液。
靶基因质粒DNA的准备用于转染的质粒DNA必须无蛋白质,无RNA和其了化学物质的污染,OD260/280比值应在1.8以上。应用酚-氯仿抽提法制备的质粒DNA一般难以达到此标准,目前大多采用进口的术提取纯化试剂盒。
具体的基因转染技术有鳞酸钙介导的转染法、DEAE葡聚糖介导转染法、脂质体介导转染法及电击基因转导尘等。靶基因被导入细胞后,一般在转染后48小时,靶基因即在细胞内表达。根据不同的实验目的,48小时后即可进行靶基因表达的检测等实验。如若建立稳定的细胞系,则可对靶细胞进行筛选,根据不同基因载体中所含有的抗性标志选用相应的药物,最常用的直核表达基因载体的标志物有潮霉素(hygromycin)和新霉素(neomycin)。
转基因动物的建立和应用
转基因动物是以实验方法交源基因导入宿主受精卵或早期胚胎细胞染色体基因组内,使其稳定整合和遗传给后代动物。它主要采用显微注射法、逆转录病毒载体感染法、精子载体法、电转移法与胚胎干细胞法。
转基因动物模型的建立,推动了肿瘤在分子水平上的研究,它使人们认识到激活的原癌基因的异常表达及功能失常,是肿瘤发生的初阶段。将癌基因与特定细胞的调控序列连在一起,可使癌基因在特定细胞中表达,研究靶细胞对不同癌基因的易感性,阐明某一癌基因对特定细胞生长、分化及功能的影响;它还可以研究多种基因的协同作用,有助于对多步骤致癌机进行深和的探讨。
转基因小鼠不但用以研究癌基因的功能,还可以通过使某一基因功能丢失(如用基因剔除技术)研究抑癌基因在肿瘤发一中的作用。另外,转基因小鼠对致癌原的测试,为肿瘤的预防,治疗及发现新的致癌物质都提供了有价值研究工具。
基因突变的检测方法
基因突变的研已成为当今生命科学研究的热点之一,检测方法也随之迅速发展。人类细胞癌基因的突变类型已如上所述,对于基因突变的检测,1985以前,利用Southern印迹法,可以筛选出基因的缺失、插入和移码重组等突变形式。对于用该法法不能检测的突变,只能应用复杂费时的DNA序列测定分析法。多聚酶链反应(polymerasechainreaction,PCR)技术是突变研究中的最重大进展,使基因突变检测技术有了长足的发展,目前几乎所有的基因突变检测的分子诊断技术都是建立于PCR的基础之上,并且由PCR衍生出的新方法不断出现,目前已达二十余种,自动化程度也愈来愈高,分析时间大大缩短,分析结果的准确性也有很大很提高。其中包括单链构象多态性(single-strandcomformational polymorphism,SSCP)和异源双链分析法(heteroduplexanalysis,HA)。下面分别介绍几种PCR衍生技术及经典突变检测方法,可根据检测目的和实验室条件选择时参考。
PCR-SSCP法PCR-SSCP法是在非这性聚丙烯酰胺凝胶上,短的单链DNA和RNA分子依其大街基序列不同而形成不同构象,一个碱基的改变将影响其构象而导致其在凝胶上的移动速度改变。其基本原理为单链DNA在中性条件下会形成二级结构,这种二级结构依赖于其碱基组成,即使一个碱基的不同,也会形成不同的二级结构而出刺同的迁移率。由于该法简单快速,因而被广泛用于未知基因突变的检测。用PCR-SSCP法检测小于200bp的PCR产物时,突变检出率可达70%-95%,片段大于400bp时,检出率仅为50%左右,该法可能会存在1%的假阳性率。应用PCR-SSCP法应注意电泳的最佳条件,一般突变类型对检测的灵敏度无大的影响,同时该法不能测定突变的准确位点,还需通过序列分析来确定。Sarkar等认为对于大于200bp的片段,用其RNA分子来做SSCP会提高其录敏度。应用PCR-SSCP检测点突变已见报道于人类大部分的肿瘤组织或细胞,如乳腺癌、食管癌、肺癌、胃癌、肝癌、胰腺癌等。检测的基因包括多种癌基因及抑癌基因,也是检测抑癌基因p53突变最常用的方法,仅检测第5-8外显子即可发现85%以上的p53基因突变。由于该法简便快速,特别适合大样本基因突变研究的筛选工作。
异源双链分析法(HA)HA法直接在变性凝胶上分离杂交的突变型一野生型DNA双链。由于突变和野生型DNA形成的异源杂合双链DNA在其错配处会形成一突起,在非变性凝胶中电泳时,会产生与相应的同源双DNA不同的迁移率。该法与SSCP相似,所不同的是SSCP分离的是单链DNA,HA法分离的是双链DNA,也只适合于小片段的分析。但HA对一些不能用SSCP检出的突变有互补作用,两者结合使用,可使突变检出率提高到近100%。
突变体富集PCR法(mutant-enrichedPCR)本法的基本原理是利用ras基因家族某个密码子部位存在已知的限制性内切酶位点,如K-ras基因第12密码子的BstNI位点,第13密古巴子有BgⅠⅡ位点。用链续二次的巢式PCR来扩增包括K-ras第12、13密码子的DNA片段,在两次扩增反应之间用相应的内切酶消化扩增的DNA片段,野生型因被酶切而不能进入第二次PCR扩增,而突变型则能完整进入第二次PCR扩增并得到产物的富集。
变性梯度凝胶电泳法(denaturing gradinent electrophoresis,DGGE)DGGE法分析PCR产物,如果突变发生在最先解链的DNA区域,检出率可达100%,检测片段可达1kb,最适围为100bp-500bp。基本原理基于当双链DNA在变性梯度凝胶中进行到与DNA变性湿度一致的凝胶位置时,DNA发生部分解链,电泳适移率下降,当解链的DNA链中有一个碱基改变时,会在不同的时间发生解链,因影响电泳速度变化的程
而被分离。由于本法是利用温度和梯度凝胶迁移率来检测,需要一套专用的电泳装置,合成的PCR引物最好在5`末端加一段40bp-50bp的GC夹,以利于检测发生于高熔点区的突变。在DGGE的基础上,又发展了用湿度梯度代替化学变性剂的TGGE法(温度梯度凝胶电泳temperaturegradientgelelectrophoresis,TGGE)。DGGE和TGGE均有商品化的电泳装置,该法一经建立,操作也较简便,适合于大样本的检测筛选。
化学切割错配法(chemical cleavage ofmismatch,CCM)CCM为在Maxam-Gilbert测序法的基础上发展的一项检测突变的技术,其检测突变的准确性可与DNA测序相仿。其基本原理为将待测含DNA片段与相应的野生型DNA片段或DNA和RNA片段混俣变性杂交,在异源杂合的双链核酸分子中,错配的C能被羟胺或哌啶切割,错配的T能被四氧化饿切割,经变性凝胶电泳即可确定是否存在突变。该法检出率很高,也是检片段最长的方法,已有报功检测了1.7kb片段,如果同时对正、反义链进行分析,检出率可达100%。应用荧光检测系统可增强敏感度,可检测到10个细胞中的1个突变细胞。该法中的化学试剂有毒,又发展了碳二亚胺检测法(catodiimide,CDI),CDI为无毒物质,也可检测大片段DNA的点突变。
等位基因特异性寡核苷酸分析法(allele-specific oligonucleotide,ASO)ASO为一种以杂交为基础对已知突变的检测技术。以PCR和ASO相结合,设计一段20bp左右的寡核苷酸片段,其中包含了发生突变的部位,以此为探针,与固定在膜上的经PCR拉增的样品DNA杂交。可以用各种突变类型的寡核苷酸探针,同时以野生型探针为对照,如出现阳性杂交带,则表运河样品中存在与该ASO探针相应的点突变,ASO需严格控制杂交条件和设置标准对照避免假阳性和假阴性。目前已有商品化的检测盒检测部分癌基因ASO突变。
DNA芯片技术(DNA chip)DNA芯片技术是90年代后发展的一项DNA分析新技术,它集合了集成电路计算机、激光共聚焦扫描、荧光标记探针和DNA合成等先进技术。可用于基因定位、DNA测序、物理图谱和遗传图谱的构建等。在基因突变检测方面DNA芯片也有广阔的前景,其基本原理为将许多已知序列的寡核苷酸DNA排列在1块集成电路板上,彼此之间重叠1个碱基,并覆盖全部所需检测的基因,将荧光标记的正常DNA和突变DNA发别与2块DNA芯片杂交,由于至少存在1个碱基的差异,正常和突变的DNA将会得到不同的杂交图谱,经过共聚集显微镜分别检测两种DNA分子产生的荧光信号,即可确定是否存在突变,该方法快速简单、片动化程度高,具有很大的发展潜力,将在基因突变检测中心发挥非常重要的作用。
连接酶链反应(ligase chain reaction,LCR)与其他核酸扩增技术比较,其最大特点为可准确区分基因序列中单个基因突变,由Landegree于1988年首次应用于镰刀奖细胞贫血的分子诊断。LCR是以DNA连接酶将某一DNA链的5`-磷酸与另一相邻链3`-羟基连接为基础,应用两对互补的引物,双链DNA经加热变性后,两对引物分别与模板复性,若完全互补,则在连接酶的作用下,使相邻两引物的5`-磷酸与3`-羟基形成磷酸二酯二酯键而连接,前一次的连接产物又作为下一次循环反应的模板,如果配对的碱基存在突变则不能连接和扩增。LCR产物检测最初是通过这32p标记上游引物3`未端,经变性凝胶电泳分离后放射自显影加以鉴定,其检测敏感性达到200个靶分子。也可设计1个横跨两引物的检测探针,用它与LCR产物进行杂交检测。近年有应用荧光素、地高辛等非核素标记方法。Batt在1994年发展了一种更为简的方法,好微孔板夹心杂交法。由于LCR的快速、特蛋和敏感的特性,以及能检测单个碱基突变的能力,因此被应用于肿瘤基因突变的分子诊断,并与PCR结合用以提高其敏感性。
等位基因特异性扩增法(Allele-specificamplification,ASA)ASA于1989年建立,是PCR技术应用的发展,也称扩增阻碍突变系统(amplificationrefractory mutation system,ARMS)、等位基因特性PCR(allele-specificPCR,ASPCR)等,用于对已知突变基因进行检测。该法通过设计两个5`端引物,一个与正常DNA互补,一个与突变DNA互补,对于纯合性突变,分别加入这两种引物及3`端引物进行两个平行PCR,吸有与突变DNA完互补的引物才可延伸并得到PCR扩增产物。如果错配位于引物的3`端则导致PCR不能延伸,则称为ARMS。ARMS和ASPCR借鉴多重PCR原理,可在同一系统中同时检测两种或多种等位基因突变位点。ASA法的检出率依赖于反应条件的优化和可能发生的引物与靶DNA有氏配时错配延伸,特别是当错配碱基为G:T时,这时可通过调整实验条件如引物靶DNA,TaqDNA聚合酶的浓度等来得高瓜在特异性。在反应体系中加入甲酰胺也可减少非特异性扩增。还可通过在引物3`端的第二个碱基引入一个错配碱基,使之与模板之间形成双重错配以阻止错误延伸。
RNA酶A切割法(RNase A cleavage)在一定条件下,氨基源双链核酸分子RNA:RNA或RNA:DNA中的错配碱基可被RNaseA切割,切割产物可通过变性凝胶电泳分离。当RNA探针上错配的碱基为嘌呤时,RNaseA在错配处的切割效率很低,甚至不切割,而当错配碱基为嘧啶时,则其切割效率较高。故如果仅分析被检DNA的一个条链,突变检出率只有30%,如同时分析正义和反义二条链,检出率可达70%。该法需要制备RNA探针,增加了操作的复杂性,但可用于1-2kb的大片段进行检测,并能确定突变位点。于这些优越性,它仍被作为一种经典方法用于对未知突变进行分析。
染色体原位杂交(In situ hybridization ofchromosome)染色体发现距今已有150多年的历史,染色体检测被广泛用于动、植物及人类的细胞遗传学研究,随着染色体分技术和分子生物学技术的发展。染色体研究范围也不断扩大,特别是用于肿瘤分子诊断。肿瘤细胞的染色体变化是一非常普遍的现象,可分为原发和继发两类。在肿瘤形成的生物学基础方面,原发性的染色体变化与引起肿瘤的直接原因有关,肿瘤细胞中可以发现各种形式的染色体畸变,如缺失、重复、易位、重排、单体断裂及核内复制等;继发性变化主要是肿瘤细胞核型的改变。染色体的检测对于肿瘤的诊断、鉴别诊断、生物学行为判别等方面都重要意义。染色体的检测方法进展很快,检测的精确率也不断提高,这里主要介绍荧光原位杂交和PRINS法。
荧光原位杂交技术(fluorescent in situhybridiaation,FISH)创建于1986年。1969年Gall和Pardue首先应用核素标记核苷酸制备探针,通过放射自显影检测杂交信号。应用核素标记的探针其敏感性可以检测到中期染色体上几百
碱基的单拷贝靶核苷酸序列,敏感性虽高,但定位不够精确。FISH具有探针稳定、操作安全,可快速、多色显示多个不同探针的杂交信号等优点。FISH的灵敏感与探针标记方法和检测仪器性能有关,探针标记时掺入的修饰核苷酸比例直接影响杂交信号强度。FISH探针一般采用随机引物法或切口翻译法,如将PCR技术引入FISH探针标记,可使其灵敏度提高到0.25kb。应用慢扫描CCD配合影像处理理软件,增强信噪比,有利于检测微弱信号,如应用TSA系统(tyramidesignalamplification)能将杂交信号再放大1000倍,可用于单拷贝基因的定位。FISH分辨率大约为1-3Mb,如果应用强变性剂处理染色体,让DNA分子从蛋白质中分离出来,使双DNA完全伸展并粘附在玻片上,经引处理后,分辨率可达1-2kb。还可采用对分裂中期染色体进行显微解剖(microdissect)法以提高分辨率。FISH的另一个特点是可以联合庆用地高辛、生物素等多种标记系统,在一次杂交中可检测多种探针在染色体上的位置及探针间的相互关系,即多色FISH或多靶FISH。FISH技术已被广泛应用于肿瘤研究中的基因扩增、易位重排及缺失等的检测,在肿瘤诊断和鉴别诊断、预后和治疗监控等方面都有重要意义。
Klch等于1989年发明了染色体上寡核苷酸引物原位DNA合成技术(oligonucleotide primed in situDNAsynthesis,PRINS),并成功地用于染色体特异α卫星DNA标记。其基本原理是用非标记的寡核苷酸引物同染色体上靶序列特异性地杂交,在DNA聚合酶作用下,当引物延伸时,掺入标记的核革酸可直接或间接地检量标记位点。PRINS技术的优点是不需克隆基因作探针,且由于杂交反应在前,标记在后,故非特异怀背景低。缺点是信号弱,灵敏度低,为克服这一制眯,Terkelsen等建立了重复引物原位标记技术(repeatedPRINS),重复进行PRINS反应,使信号号强度明显提高。基于FISH的原理,发展了多色原位经物标记(multicolorPRINS),可测定二个以上靶序列在DNA分子上的相互的位置,且特异性比FISH还高。
DNA序列分析(DNA sequencing)应用各种突变检测技术检测到的基因突变,最后都需用序列分析才能确定突变类型及突变位置,其效率可以达到100%。现在的测序方法已与经典的方法有了很大的不同,其基本原理虽仍是双脱氧终止法,但自动化程度大为提高,操作更简便,测序时间也大大缩短。随着PCR技术与测序联合使用,不需经过M13亚克隆步骤,故称为直接测序法(directsequencing,DS)。DS法测序的模板主主要来源于PCR,应用不对称PCR(asymmetricPCR)和基因组扩增转录同步测序法(genomic amplification with transcriptsequencing,GAWTS)等,使单链产物大大增加。近年来,PCR循环测序法的建立,使模板扩增与同步进行,引物用四种不同前颜色的荧光标记,使每个样品的四个测序反应可在一个反应管和一个泳道内进行,大大提高了测邓的自动化程度。目前PE公司推同出的DNA自动测序仪已发展到96泳道,并仍在不断改进。这些高度自动化的测序方法是经较理想的基因突变分析技术,但其昂贵的费用其使用范围,所以对一些小样本或为了某些特定目的的样本分析,仍进行经典的手工测序。
突变基因的检测方法多种多样,特别是PCR技术诞生后,许多检测技术都是在PCR基础上衍生的。由于PCR扩增南非要的模板量少,使对肿瘤的突变分析可以精确到单细胞,如霍奇金病的R-S细胞的单细胞基因突变分析。另外,应用显微解剖法(microdissection)可在组织切片上较精确地挑选需检测的靶点,其优点是可克服肿瘤组只中间质或癌周组织的混杂,提高准确性。除上述介绍的方法外,还有多种方法也用于基因突变的分子检测,如对未知突变基因进行分析的酶促切割错配法(enzymemismatc cleavage,EMC)、切割片段长度多态性(cleavage fragment length polymorphism,CEFLP)、双脱氧指纹图谱法(dideoxy fingerprinting,ddF)、错配接合蛋白质截短测试法(protein truncationtest,PTT)等。对已知突变基因进行分析 的有引物延伸法(primerextension,PEX)、寡核苷酸链接检测法(oligonucleotide ligationassay,OLA)、毛细管电泳法(capillary electrophoresis,CE)等方法。
细胞生物学实验方法与技术
当前细胞生物学与医药保健事业联系的较为紧密的热点问题主要有以下几种:1)真核细胞基因结构及其表达调控;2)细胞膜、膜系、受体与信号传递研究;3)细胞生长、分化、衰老、癌变、死亡,尤其是程序性细胞死亡的研究;4)细胞工程,包括基因工程及体细胞核移植的研究。
一、细胞培养常用方法
1、细胞原代培养(primay culture)又称初代培养,即直接从机体取下细胞、组织、或器官、让他们在体外维持与生长。原代细胞的特点是细胞或组织刚离开机体,他们的生物状态尚未发生很大的改变,一定程度上可反映 他们在体内的状态,表现出来源组织或细胞的特性,因此用于药物实验尤其是药物对细胞活动、结构、代谢、有无毒性或杀伤作用等研究是极好工具。常用的原代培养方法有组织快培养法及消化培养法。前者方法简单,细胞也较易生长,尤其是培养心肌有时能观察到心肌组织块的搏动。细胞从组织块外长并铺满培养皿或培养瓶后即可进行传代。2、细胞的传代培养当细胞生长至单层汇合时,便需要进行分离培养否则会因无繁殖空间、营养耗竭而影响生长,甚至整片细胞脱离基质悬浮起来直至死亡。为此当细胞达到一定密度时必须传代或再次培养,目的是借此繁殖更多的细胞,另一方面是防止细胞的退化死亡。
二、器官培养方法
器官培养(organculture)是指用特殊的装置使器官、器官原基或它们的一部分在体外存活,幷保持其原有的结构和功能。器官培养可模拟体内的三维结构,用于观察组织间的相互反应、组织与细胞的分化以及外界因子包括药物对组织细胞的作用。
器官培养方法很多,最经典的方法即表玻皿器官培养法;一种最常用的方法是不锈钢金属网格法及Wolff培养法和扩散盒培养法,实验者可根据情况选择采用。
三、放射自显影术测定
放射自显影术(autoradiography)是利用放射性同位素电离辐射对核子乳胶的感光作用,显示标本或样品中放射物的分布、定量以及定位的方法。放射性同位素能在紧密接触的感光乳胶中记录下它存在的部位和强度,准确显示出形态与功能的定位关系。现已可将放射自显影术与电镜以及生物分子结合起来。不但可以研究放射性物质在组织和细胞内的分布代谢,而且可以揭示核酸合成及其损伤等改变,目前已在生命科学各领域被广泛应用。
四、染色体分析技术
染色质或染色体是遗传物质在细胞水平的形态特征。前者是指当细胞处于合成期时遗传物质经碱性染料着色后,呈现出细丝状弥漫结构;当细胞进入分裂期时,染色质细丝高度螺旋化凝聚为形态有特征的染色体。特别是在分裂中期,复制后的染色体达到最高程度的凝聚,称为中期染色,是进行染色体形态观察分析的最佳时期。染色体分析应用领域越来越广,主要用于以下几方面:1)为临床诊断提供新手段;2)研究不育和习惯性流产发生的遗传基础;3)通过检查胎儿的染色体,预防有染色体异常患儿出生(先天愚型);4)根据染色体的多肽性进行亲子和异型配子的起源研究;结合DNA重组技术可以将基因定位于染色体的具体区带上。
五、电镜技术
早在1940年,英国剑桥大学首先试制成功扫描电子显微镜,但因分辨率低无实用价值。1965年英国剑桥科学仪器有限公司开始生产出商品扫描电镜,其以显著优点广泛用于生物学、医学、物理学、化学、电子学及勘探、冶金、国防、公安、机械与轻工业等诸多领域,并已成为非常有用的研究工具。
电镜主要特点:1)景深大,较光学显微镜大几百倍;2)图像富有立体感,是一个具有真实感的三维结构立体图象;3)图像放大范围大,光学显微镜有效放大倍数为1000倍左右,透视电镜的放大倍数为几百倍至100万倍,扫描电镜可放大十几倍至几十万倍;4)分辨率高,扫描电镜可达6-3nm;5)样品可在三度空间平移和旋转,聚焦后可以任意放大倍数,而不需调整重新聚焦。
六、细胞、细胞器、及细胞间质的分离技术、
1、 细胞的分离分离不同的细胞及亚细胞组分在现代生物学研究中起着重要的作用。如研究某种药物治疗白血病的机理,需要分离培养人或动物的骨髓细胞,观察药物的细胞作用;研究与细胞生长分化有关的生长因子的作用,需将与此类因子有关的细胞分离出来;分离细胞膜,线粒体等细胞的亚组分,对于研究信号传递,某些遗传疾病,也都是必不可少的手段。
2、细胞膜的分离细胞内的膜系统与细胞质膜统称为生物膜(biomembrane),他们都有共同结构和特征。首先要分离出形状完整的、具有生物活性的、高纯度的细胞膜,用于研究细胞膜的结构和功能,以利于观察膜在细胞与环境进行能量交换及信息传递的过程。
3、细胞核的分离细胞核作为一个功能单位,完整的保存遗传物质,幷指导RNA合成,后者为蛋白质及其它细胞组分合成所必需。因此细胞核分离是研究基因表达及细胞核形态结构的首要步骤。不同组织来源的细胞经匀浆后,用分级离心或超声波处理等方法进行纯化。
4、溶酶体的分离溶酶体是处理细胞吞噬物的细胞器,含有高浓度的各种水解酶类,调控细胞内的消化过程。溶酶体的分离常用于研究因溶酶体功能缺陷而引起的多种疾病。
5、线粒体的分离线粒体是细胞呼吸的主要场所,细胞活动所需要的能量,主要由在线粒体内进行氧化所产生的能量供给。制备线粒体关键是保持其完整性及高纯度。
6、细胞DNA、RNA分离与纯化核酸是遗传信息及基因表达的物质基础。核酸的提取与纯化关键是保持核酸的完整性,但较困难,主要因为:一是细胞内有活性很高的核糖核酸酶;二是酸碱等化学因素;三是高温机械损伤等物理因素,需严格遵守操作规程。
七、细胞凋亡研究方法
细胞凋亡(apoptosis),又称为程序性死亡(programmed celldeath,PCD)指的是有核细胞在一定条件下,通过激活其自身内部机制,尤其是开启与关闭某些基因以及内源性DNA内切酶活化,导致产生细胞自然性死亡的过程。可以认为细胞死亡的这种方式是一种生理性的自发过程。为此有人也称其为细胞自杀。
目前认为程序性死亡几乎和细胞的增殖同样重要,如果没有细胞凋亡,个体不能形成与存活,或者发生疾病。只有通过细胞凋亡的发生,使特定细胞群体在特定的时间和特定的部位死亡。从而使机体在总体上保障其细胞数量,以及形态与功能的平衡。近年来如何用药物诱导癌细胞死亡也成为细胞凋亡的热点之一。透视电镜观察是研究细胞凋亡的首选方法。
八、原位杂交
原位杂交技术是将分子杂交与组织化学相结合,用标记的DNA或RNA为探针,在原位检测组织细胞内特异互补的DNA或RNA序列。它分为三类:DNA-DNA、DNA-RNA、RNA-RNA。已广泛用于生物学的各个领域。原位DNA末端标记可以用于细胞凋亡的定量研究。原位杂交技术和PCR技术结合用于检测人乳头瘤病毒(HPV)
九、单克隆抗体
是1975年被描述的一种可产生无限量,并可预测其性质的抗体制备技术。该技术的关键步骤在于淋巴细胞之间的融合,所以称为“淋巴细胞杂交瘤技术”;由于杂交瘤经过筛选可产生针对单一抗原决定簇抗体的细胞克隆,故称为单克隆抗体技术。该技术的问世使得免疫学研究与实践发生了革命性的改观,同时还为生物学和医药学的许多领域提供了前所未有的研究工具。人们利用其可精确地识别出极为复杂的分子,测定出无法测定的物质,识别出新的细胞群体,揭示了以往未曾了解的细胞分化途径,为肿瘤、感染性疾病、自身免疫性疾病等的诊断和治疗创造出令人兴奋的前景。
十、神经细胞培养神经细胞培养是指从体内取出某一种神经组织,在无菌、适当温度和一定营养条件下模拟在体生理环境,使之存活和生长,幷保持其结构和功能。
神经细胞培养按培养前切割程度分为器官培养、组织快培养和细胞分离培养。
1、器官培养 (organ culture )是切割最少型的培养。全器官或器官大片放在保温的营养液中可保存数日。这类培养切断了正常的血供,器官的营养交换取决于简单的灌流。
2、组织块培养 (explant culture)是真正最小器官培养。组织块通常厚度为0.5-1mm,一个或几个组织块放在一个培养皿内存活数周。实验在其组织块周围生长出的边缘细胞上进行。
3、细胞分离培养 (dissciated cell culture )是由分散其原始组织至其组成的细胞。常用酶进行消化。