转载
例子,计算Cu的态密度
一、自洽计算
&control
calculation='scf'
restart_mode='from_scratch',
pseudo_dir = './',
outdir='./'
prefix='cu'
tstress = .true.
tprnfor = .true.
/
&system
ibrav = 2, celldm(1) =6.73, nat= 1, ntyp= 1,
ecutwfc = 25.0, ecutrho = 300.0
occupations='smearing', smearing='gaussian',degauss=0.02
/
&electrons
diagonalization='david'
conv_thr = 1.0e-8
mixing_beta = 0.7
/
ATOMIC_SPECIES
Cu 63.55 Cu.pz-d-rrkjus.UPF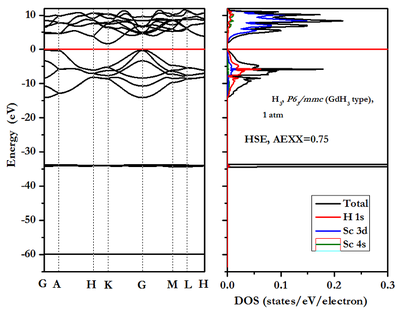
ATOMIC_POSITIONS
Cu 0.0 0.0 0.0
K_POINTS (automatic)
8 8 8 0 0 0
解释:
在电子自洽计算中需设置以下几个方面的参数:
1)控制计算的部分,也就是要设置
&control
.....
第一个'/'之间的关键词。
关键词calculation赋值为'scf'表示此计算是进行自洽电荷密度计算;
restart_mode表示是否是接着上一次的计算而继续的计算,赋值为'from_scratch'意味着是进行一次全新的计算开始;
pseudo_dir用来设置赝势文件所在的目录,赋值为'./'表示赝势文件放在当前计算目录;
outdir用来设置计算过程中输出文件(比如波函数、电荷密度以及势)输出到哪个目录中。赋值为'./'表示这些输出文件将放到当前计算目录中;
prefix用来定义当前计算作业的标题名,它将是一些主要输出文件的文件名。赋值为'cu'用来标记当前计算作业是对Cu进行计算;
tstress 用来设置在自洽计算过程中是否计算体系的应力,设置为 .true.表示在自洽计算过程中要计算体系的应力;
tprnfor 用来设置在自洽计算过程中是否计算体系中原子所受的力,设置为.true.表示在自洽计算过程中要计算体系中原子所受的力;
2)、描述所计算的体系(包括它的晶格类型、晶格常数或结构参数、原胞基矢、原胞中原子的类型数目和总的原子数目)、平面波的切断动能(也就是在展开KS轨道或晶体波函数的平面波切断动能;另外,还包括在计算电荷密度时,展开的平面波的切断动能)、确定电子占有数的方法及相关的参数。也就是由
&system
..........
第二'/'之间的关键词来设置。
ibrav用来归属体系所属的晶格类型,赋值为2表示所计算的体系是fcc结构;
celldm(1)用来设置体系的第一个晶格常数,因为所计算的体系是fcc结构,只需设置celldm(1),相当于指定晶格常数a的值;
nat用来指明体系的原胞中原子的总共数目,赋值为1表示所计算的原胞中只有一个原子;
ntyp用来指明体系中原子类型的数目,赋值为1表示所计算的体系只有一种类型的原子;
occupations用来设置确定电子占有数的方法,赋值为'smearing'表示采用smearing的方法来确定电子的占有数,随后须设置smearing和degauss关键词;
smearing用来指明确定电子占有数的一种具体的smearing方法,赋值为'gaussian'表示采用Gaussian函数来确定电子占有数;
degauss用来确定smearing方法中有关函数的展宽参数,赋值为0.02表示上面Gaussian函数中的展宽参数为0.02。
3)、设置电子自洽计算中本征矢量(波函数)和本征值的计算算法,自洽收敛的标准。也就是
&electrons
......
和第三个'/'之间的关键词来设置。
diagonalization用来设置在求KS方程的本征矢量和本征值时,采用具体的什么算法,赋值为'david'表示采用Davidsoniterative diagonalization with overlapmatrix方法;
conv_thr用来设置自洽收敛标准,赋值为自洽循环过程总能的变化小于1.0e-8的化,那自洽计算就停止;
mixing_beta用来设置自洽计算过程中前后两次电荷密度混合的参数。
4)、指明体系中原子的元素名,原子量以及所采用的赝势,即ATOMIC_SPECIES后面的设置,它们的顺序要和后面原子的坐标一一对应起来。
Cu 63.55 Cu.pz-d-rrkjus.UPF
表示所计算的体系中原子是Cu,它的原子量为63.55,它的赝势文件为Cu.pz-d-rrkjus.UPF。
5)、给出体系原胞中原子的坐标位置,也就是ATOMIC_POSITIONS 后面的设置:
Cu 0.0 0.0 0.0
表示原胞中第一个原子是Cu,它位于原胞的原点。
6)、k点取样的设置,也就是K_POINTS 后面的设置:
K_POINTS (automatic)表示由程序采用M-P方法自动确定k点,需给出k点取样网格的大小,以及是否在产生k点后对这些点进行平移。
8 8 8 0 0 0
表示采用8x8x8的网格来确定k点,而且不对k点进行平移。
二、非自洽计算,增加k点,并采用四面体方法来确定电子的占有数
&control
calculation='nscf'
prefix='cu',
pseudo_dir= './',
outdir='./'
/
&system
ibrav=2,celldm(1) =6.73, nat=1, ntyp=1,
ecutwfc= 25.0, ecutrho = 300, nbnd=8,occupations='tetrahedra'
/
#[【非自洽计算中应指定nbnd,那么在自洽计算中有必要指定nbnd数量吗?如何在结果文件中查看实际使用的nband的数量。每个k点对应的能量特征值,是不是就是band(或level, bands consists of levels,consists of band ;levels formband)】
&electrons
conv_thr= 1.0e-8
mixing_beta= 0.7
/
ATOMIC_SPECIES
Cu63.55 Cu.pz-d-rrkjus.UPF
ATOMIC_POSITIONS
Cu 0.00.0 0.0
K_POINTS{automatic}
12 1212 0 0 0
三、采用dos.x计算总态密度
&inputpp
outdir='./'
prefix='cu'
fildos='cu.dos',
Emin=-5.0,Emax=25.0, DeltaE=0.1
/
四、采用projwfc.x来计算分波态密度
&inputpp
outdir='./'
prefix='cu'
Emin=-5.0,Emax=25.0, DeltaE=0.1 ngauss=1,degauss=0.02
/
在计算态密度的步骤就是如上面所述:a),先进行自洽计算,保留输出的势、电荷密度和波函数;b),然后读入上一步自洽计算得到的势或电荷密度或波函数,进行非自洽计算,其中增加k点网格,并采用四面体方法来确定电子占有数;c),采用dos.x计算总态密度;d),采用projwfc.x计算分波态密度。
【分态密度的英文原意为Atomic orbital projected density ofstates,即按照不同原子往s p d不同的轨道上投影态密度。projwfc.x操作的英文释义:projectswavefunctions onto orthogonalized atomic wavefunctions, calculates Lowdin charges, spilling parameter,projected DOS (separated into up and downcomponents for lSDA) alternatively, computes thelocal DOS(E), integrated in volumes given ininput】
在自洽计算中occupations关键词的赋值已设置为'tetrahedra'表示采用四面体方法确定电子占有数和费米能级。另外,K_POINTS{automatic}下面的k点设置已增密,设置为12x12x12,为使得计算的态密度光滑,有可能需设置的更密些。
#[【非自洽计算中应指定nbnd,那么在自洽计算中有必要指定nbnd数量吗?如何在结果文件中查看实际使用的nband的数量。每个k点对应的能量特征值,是不是就是band(或level, bands consists of levels,consists of band ;levels form band)】
在采用dos.x总态密度计算中,输入文件中由&inputpp和'/'来之前的关键词来设置,它的关键词有:
outdir用来设置计算上非自洽计算输出文件的目录,设置为'./'表示是当前目录;
prefix用来标记当前所计算的体系,也确定了上一步非自洽计算输出的势或电荷密度或波函数的文件的名称,此例子中设置为'cu',注意它们的赋值应该与上一步的非自洽计算中的一致。
fildos用来指明所计算的总态密度将写到哪个文件中,此例子中赋值为'cu.dos',表示总态密度将写到cu.dos文件;
Emin用来设置计算态密度时,能量范围的最小值,赋值为-5.0,表示将从E=-5.0eV开始输出对应的态密度值;
Emax用来设置计算态密度时,能量范围的最大值,赋值为25.0,表示将到E=25.0eV为止输出对应的态密度;
DeltaE用来设置计算态密度时,按多大的能量间隔输出态密度,这里设为0.1eV输出态密度。
在采用projwfc.x计算态密度时,&inputpp和'/'来之前的关键词来设置,它的关键词与dos.x的输入文件中的关键词差不多:
ngauss用来设置态密度时展宽的方法,这样是为了使得所计算的态密度看起来光滑,可以赋值:
0,表示采用简单的高斯函数【默认值为0】
1,表示采用一阶Methfessel-Paxton函数
-1,表示采用Marzari-Vanderbilt“冷离散“方法,其实就是一种函数形式
-99,表示采用Fermi-Dirac函数
degauss用来设置展宽函数中的展宽系数
仿照上述例子写的Sc在1atm压力下的态密度计算完整脚本为(a指压力):
#!/bin/sh
####################################################################
PW_ROOT=/home/pwscf/pwscf/espresso-4.0.5/bin/
PSEUDO_DIR=/home/workplace/pwscf-phononpy/pseudo
TMP_DIR=/home/workplace/pwscf-phononpy/tmp
#export PARA_PREFIX='mpirun -np 2'
# or export PARA_PREFIX='mpirun' ,exportPARA_POSTFIX= -np 3
export PW_ROOT PSEUDO_DIR TMP_DIR
export name='Sc' #【定义工作环境,赝势及命令存放位置,临时文件存放位置等】
############################################################
for a in 0
do
cat > name.scf.in_a <<EOF
&control
calculation = 'scf'
restart_mode='from_scratch',
prefix='$name',
pseudo_dir ='$PSEUDO_DIR/',
outdir='$TMP_DIR/'
tstress=.t.,
tprnfor=.t.
/
&system
ibrav=0,
nat=2,
ntyp=1,
ecutwfc=90.0, ecutrho =360.0
occupations='smearing',
smearing='methfessel-paxton',
degauss=0.05
/
&electrons
conv_thr = 1.0d-8
mixing_beta = 0.7
/
#&CELL
#cell_dynamics= 'damp-pr' #【自洽计算这些参数都没有必要,但为了完整性还是写出来】
# press=$a
#/
ATOMIC_SPECIES
Sc 44.9559 Sc.pbe-nsp-van.UPF
CELL_PARAMETERS {bohr}
6.254820382 0.000000000.00000000
-3.127410191 5.416833340.00000000
0.000000000 0.000000009.958412336
ATOMIC_POSITIONS {crystal}
Sc 0.3333333333333286 0.6666666666666714 0.2500000000000000
Sc 0.6666666666666714 0.3333333333333286 0.7500000000000000
K_POINTS {automatic}
8 8 4 0 0 0
EOF
done
##################################################################################################
cat > $name.nscf.in << EOF
&control
calculation='nscf',
prefix='$name'
pseudo_dir = '$PSEUDO_DIR/',
outdir='$TMP_DIR/'
/
&system
ibrav = 0,
nat= 2,
ntyp= 1,
ecutwfc = 90,
ecutrho = 400.00,
occupations='smearing',
smearing='methfessel-paxton',
degauss=0.05
nband=16
/
&electrons
mixing_beta = 0.7
conv_thr = 1.0d-8
/
#&CELL
#cell_dynamics= 'damp-pr' #【自洽计算这些参数都没有必要,但为了完整性还是写出来】
# press=$a
#/
ATOMIC_SPECIES
Sc 44.9559 Sc.pbe-nsp-van.UPF
CELL_PARAMETERS {bohr}
6.254820382 0.000000000.00000000
-3.127410191 5.416833340.00000000
0.000000000 0.000000009.958412336
ATOMIC_POSITIONS {crystal}
Sc 0.3333333333333286 0.6666666666666714 0.2500000000000000
Sc 0.6666666666666714 0.3333333333333286 0.7500000000000000
K_POINTS { automatic }
12 12 6 0 0 0
EOF
######################################################################################################
cat > $name.Dos.in << EOF
&inputpp
prefix = '$name'
outdir = '$TMP_DIR/'
fildos = '$name_dos.dat'
DeltaE=0.01
/
EO F
####################################################
cat >$name.pdos.in << EOF
&inputpp
outdir='$TMP_DIR/'
prefix='$name'
DeltaE=0.01
/
EOF
###################################################################