脊髓栓系综合征(TCS)是由多种原因引起的,脊髓、神经被非弹性结构固定,在生长发育过程中圆锥被固定在低位,导致脊髓、神经损伤,并由此产生一些列神经功能障碍的综合征。而圆锥位置正常的TCS为隐匿性TCS。TCS的病因包括基因和染色体的异常、胚胎发育的异常、后天原因等。
一些作者通过动物实验和人类染色体与基因的研究表明,TCS与某些基因和染色体的变异有关。但是关于TCS的染色体和基因的研究并不多,而这些研究中大部分是病例报道,HLXB9基因突变的患者多伴有椎体和肛门的畸形,有60.87%HLXB9基因突变的患者被诊断为TCS,而这些患者中只有17.39%的TCS患者没有神经管缺损(NTD)。
Bassuk等人通过对4个家族的研究发现许多TCS患者都存在基因学的基础:22q11.2缺失可能会导致初级神经胚形成障碍,一些22q11.2上的TBX1基因发生错意突变的22q缺失综合征患者合并有TCS,说明TBX1基因可能是TCS相关基因,许多22q缺失综合征的儿童会出现严重的步态异常、便秘、小便失禁或者行走迟缓等神经损伤表现,而22q11.2缺失可以表现为不完全的外显率和多样的表现度,所以确认为22q11.2缺失的患者出现轻微的神经损伤表现的主要原因可能合并有TCS。
21三体的母亲所生的孩子合并有神经管缺损的风险更大,21三体综合征的患者如果有TCS神经损伤的表现应该考虑TCS的可能。环状22号染色体、13q32三体、8三体和NF1也可能是导致TCS的异常变异,所以对于任何染色体异常的患者都应考虑是否合并有TCS。说明一些由于基因或染色体异常引起的TCS可能存在遗传性。
胚胎发育的异常
脊髓和椎管分别由胚胎时期的外胚层和中胚层发育而来,妊娠第18~28d开始外胚层增生形成神经板,并逐渐闭合形成神经管,第11周时骨性椎管闭合,第12周时脊髓延伸于整个椎管,其尾端和椎管末端相平,但在生长发育的过程中椎体生长发育的速度,即椎管增长的速度比脊髓增长的速度要快,由于头端是固定的,故逐渐出现脊髓向头侧“上移”的现象,一般圆锥上升开始于妊娠后第43~48周,每个月大约上移半个椎体水平,第4个月脊髓已达到骶椎以上,第5个月位于腰椎下端,第6个月脊髓圆锥到达L4、5椎体水平,25~33周时脊髓圆锥位于L1~3水平,出生时圆锥位于L1、2水平,但脊髓位于L2、3水平以上也认为是正常的,出生后脊髓继续移向头端,98%足月儿和非足月儿出生后1周圆锥达到L2水平,出生后1~6个月脊髓圆锥末端逐渐达到成人水平,即T12~L1椎体下缘水平(大约5%的成年人的圆锥位于L2水平),一般上下不超过25px的范围。
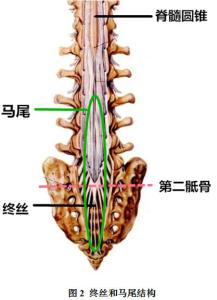
如果在初级神经胚形成时有间充质细胞进入神经管的尾端或者在二级神经胚形成时有尾端细胞团块进入后神经孔,都会造成脊柱裂或者皮毛窦。脊髓圆锥尾部的细胞团退化与软脊膜共同形成终丝(成人终丝直径一般不超过2mm),附着于第1~2尾骨膜背侧,终丝有固定脊髓的作用,从尾椎至S2水平长约5~200px的终丝位于硬膜囊下端,称为外终丝;S2水平以上长约375px的终丝位于硬膜囊内,成为内终丝。
正常柔软纤细的终丝和齿状韧带对牵拉具有一定的缓冲作用,允许脊髓在生长发育过程中逐渐上移,但如果终丝变短增粗后,这种缓冲作用就减弱甚至消失了,当脊髓神经长期被过度牵拉,其血流、代谢和电生理功能等方面发生改变,进而出现一系列的TCS神经功能损害的临床表现。发育异常导致的TCS的病因包括脊柱发育畸形、椎管闭合不全、椎管内脂肪瘤、皮样囊肿、脊髓脊膜膨出、脂肪性脊髓脊膜膨出、脊髓发育畸形等。
氧化损伤机制
20世纪80年代到90年代的一些学者通过动物实验发现,缺氧、脊髓血供障碍、脊髓圆锥受到牵拉后还原型细胞色素α1,α3增加,即脊髓末端线粒体代谢率有所降低,而且脊髓圆锥的电位也随之降低或消失。其张力越大、作用时间越长,还原型细胞色素α1,α3水平越高,脊髓圆锥电位越低,即脊髓圆锥氧化代谢水平越低,而且其血流也会降低,神经损害也越严重,在脊髓受到牵拉后,其线粒体的氧化代谢功能下降,进而导致感觉和运动神经功能的损害。