【关键词】尘埃粒子计数器 洁净室 在线监测 风险评估
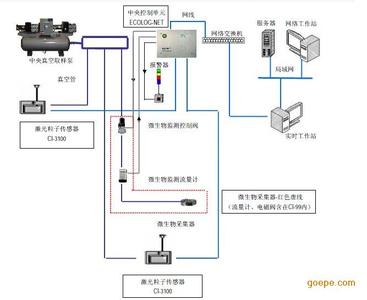
为了满足GMP规范,用于制药生产的洁净室需要符合相应的等级要求。所以这些无菌生产的环境需要严格的监测以确保生产过程的可控性。需要进行重点监测的环境一般安装一套尘埃粒子监测系统,该系统包括:控制界面、控制设备、粒子计数器、气管、真空系统和软件等。在每一个关键区域都装有一个连续测量的粒子计数器,通过工作站电脑激发指令对每个区域进行连续的监测采样,监测的数据传给工作站电脑,电脑接收到这些数据可以显示和出具报告给操作者。尘埃粒子在线动态监测位置、数量的选择都应基于风险评估研究,要求覆盖到所有的关键区。
1 粒子监测的规则
所有使用无菌工艺生产的无菌药品和生物制品必须遵循现行版药品生产质量管理规范(GMP)。在美国这些规范由美国食品药品监督管理局(FDA)制定为21号联邦法规(21 CFR)的210和211部分。在欧盟这些规范由欧盟制定为药品生产质量管理规范,附录1“无菌药品生产”。 由ISO/TC209洁净室及相关受控环境计数委员会提出的ISO 14644-1是洁净室污染等级定义/分级的国际标准。制药公司生产的产品因此必须证明药品在发行到市场及最终用户之前的每个步骤都是遵从法规来执行的。
为满足所需的要求,确保在一个可控的环境中生产产品。洁净室的建立主要用于污染控制,减少潜在生产环境的变化,洁净室的设计必须符合相应的洁净度要求,包括达到“静态”和“动态”的标准。GMP规范要求,同时针对于微生物和非微生物污染,必须严格监测这些环境来确保当前环境条件的完整性及持续可见性,尽可能降低产品或所处理的物料被微粒或微生物污染的风险。
2 洁净室粒子计数的三种测量阶段
(1)建成:服务连接和服务功能完备的房间,但设施内没有生产设备和人员。
(2)静态:所有的服务已连接的条件,设备已经安装并在一个约定的方式下运行,但没有人员在场。
(3)动态:所有设备已经安装并以制定的形式运行,有指定数量的人员在场,并以制定的流程工作。
3 关键区域
在无菌原料操作区域是非常关键的,因为裸露的产品很容易受到污染,并且随后在其直接接触容器中不能被灭菌。为了保持产品的无菌性,将无菌操作环境(例如设备放置、灌装)控制并维持在适当质量水平是必要的。环境质量的一个方面是空气的微粒含量。粒子数是非常重要的,因为它们可以作为一种外源性污染物进入一个产品,也可以作为微生物的载体来产生生物污染。适当设计的空气处理系统可将关键区域的微粒含量减至最少。
FDA推荐,关键区域的空气洁净度的确认测量应在对于暴露的灭菌产品、容器和密封包最具潜在风险的位置进行。粒子计数采样头应放置于最有意义的采样位置。在每个生产周期中应定期监测。同时FDA推荐,非微生物颗粒监测使用远程计数系统。这些系统能采集更全面的数据,且比便携式粒子计数器产生更小的侵害。
粒子测量是基于使用离散空气粒子计数器来测量大于等于指定粒径的粒子浓度。一个持续测量系统应用来测量A级区域的粒子浓度,在B级围绕区域也推荐使用。对于常规测试,A级和B级区域总采样体积量不应小于1立方米。
4 监测点安装选择模拟测试
模拟实际生产过程(如药品灌装),在选定的关键区域内通过对各候选粒子采样点位的测量结果,确定尘埃粒子计数器采样头的安装位置。首先进行静态模拟测试,以证明接下来的生产(灌装)模拟测试是在一个合格的基础环境下进行的。然后进行动态生产模拟测试,将产生支持性数据,最终确定每个关键区域内的尘埃粒子采样点位。
4.1确定关键区域
列出无菌生产区域药品或灭菌容器容易暴露的高风险区(A级或B级)。对放置粒子计数器的监测点的位置、选择原因应依据实际的生产工艺进行详细的分析。其中A级区为高风险操作区,如:灌装区、放置胶塞桶和与无菌制剂直接接触的敞口包装容器的区域及无菌装配或连接操作的区域,应当用单向流操作台(罩)维持该区的环境状态。B级区通常为无菌配置和灌装等高风险操作A级洁净区所处的背景区域。对于B级区是否需要粒子监测系统,则取决于B级区和A级区之间是否存在明显的隔断。
而非关键区(C级或D级)不需要进行对尘埃粒子的连续监测。C级和D级区域指无菌药品生产过程中重要程度较低操作步骤的洁净区。因为这些区域属于低风险区,可以通过使用便携式的粒子计数器来完成日常的监测。
中国2010版GMP附录1对各级别空气尘埃粒子标准规定如表1。
表1 不同洁净级别尘埃粒子标准
洁净度级别 悬浮粒子最大允许数/立方米
静态 动态
≥0.5μm ≥5.0μm ≥0.5μm ≥5.0μm
A级 3520 20 3520 20
B级 3520 29 352000 2900
C级 352000 2900 3520000 29000
D级 3520000 29000 不作规定 不作规定
4.2静态模拟测试
在模拟动态生产过程测试进行之前应进行静态模拟测试,以确认各关键区域的环境粒子级别是否在规定的范围以内。如果测试数据表明环境粒子级别超过该区域应有的(设定的)级别,则说明该区域环境已遭到污染,应在模拟生产过程之前先解决环境污染问题。
在静态环境下,分别在各关键区域中心正上方25cm处,用采样流量为100L每分钟的便携式粒子计数器连续采集10分钟,将最后结果与中国2010版 GMP附录1的要求比对,如果结果符合该点设计等级要求的静态环境下尘埃粒子最大允许数,则此关键点的静态环境通过测试。需注意在确认时,应当使用采样管较短的便携式尘埃粒子计数器,避免≥5.0μm尘埃粒子在长采样管中沉降。 4.3动态生产模拟测试
关键区域静态环境测试合格之后,开始动态生产模拟测试。完全模拟生产过程,确认洁净室无菌工作现场的环境良好,无喷洒、清洁、消毒等影响粒子计数器运行的操作,对各关键区域中分别选择若干可行的在线粒子采样候选点位,然后使用便携式粒子计数器进行模拟测量。
对候选点位的确定建议参考以下原则:
(1)ISO 14644-1 规范说明:对于单向流洁净室,采样口应对着气流方向;对于非单向流洁净室,采样口宜朝上,采样口处的采样速度均应尽可能接近室内气流速度;
(2)GMP原则:采样头应安装在接近工作高度和产品暴露处;
(3)采样位置不会影响生产设备的正常运行,同时不会影响到生产过程中的人员正常操作,避免影响物流通道
(4)采样位置不会因产品本身产生粒子或滴液引起大的计数误差,导致测量数据超过限定值,并且不会对粒子传感器造成损害;
(5)采样位置应具备安装可能性;
(6)采样位置选在关键点水平面以上,距离关键点不应超过30cm,特殊位置如有药液飞溅或溢出,导致模拟生产状况下测量数据结果超出该等级区域标准,可将垂直方向的距离限制适当放宽,但不宜超过50cm;
(7)应尽量避免将采样位置放于容器经过的正上方,以免引起容器上方空气不足和产生扰流。
确定了所有候选点位后,在模拟生产环境的条件下,用采样流量为100L每分钟的便携式粒子计数器在各关键区域的各候选点位进行10分钟的采样,并对所有点位的尘埃粒子采样数据记录。对同一区域的多个候选点位的采样结果进行对比分析,找出高风险的监测点,从而确定该点为最终的尘埃粒子监测点采样头安装位置。
5 结语
本文主要阐述了通过一个风险评估的方法,确定洁净室在线尘埃粒子监测系统粒子计数器采样头的安装位置。首先依据生产工艺确认无菌生产厂房洁净室中的关键区域,即识别产品生产过程中容易被污染的高风险区域。然后对各个高风险区域进行静态模拟测试,确认各区域符合其应保持的洁净度级别,证明环境没有受到污染。最后在每个高风险区域分别选取若干候选点位进行动态生产模拟测试,选出环境最差点,即高风险区域的高风险监测点,将其确定为粒子计数器采样头的最终安装位置。
该方法通过一系列测试对监测点位的选择提供了数据支持,选出的尘埃粒子监测点具有代表性,通过系统连续的在线监测,能够较好的反应洁净室真实的环境状态,降低了无菌药品和生物制品被微粒或微生物污染的风险。
参考文献:
[1]药品生产质量管理规范(2010年修订)[S].附录1.
[2]ISO 14644 2007洁净室及相关受控环境国际标准[S].
[3]蒋井明.悬浮粒子在线动态监测系统的应用探析[J].机电信息.2013,(23):25-28.
[4]魏嵬.悬浮粒子在线监测系统设计实例[J].洁净与空调技术.2012,(2):71-75.




