(2) 组织中总蛋白的提取:
1、 将少量组织块置于 1~2ml 匀浆器中球状部位,用干净的剪刀将组织块尽量剪 碎。
2、 加 400μl 单去污剂裂解液裂(含 PMSF)于匀浆器中,进行匀浆。然后置冰上。 3、 几分钟后再碾一会儿再置于冰上,要重复碾几次使组织尽量碾碎(870+10)。 4、 裂解 30 min 后,即可用移液器将裂解液移至 1.5ml 离心管中,然后在 4℃下 12000rpm 离心 5min,取上清分装于 0.5ml 离心管中并置于-20℃保存。 (3) 加药物处理的贴壁细胞总蛋白的提取:
由于受药物的影响,一些细胞脱落下来,所以除按(一)操作外还应收集培养液 中的细胞。以下是培养液中细胞总蛋白的提取:
1、 将培养液倒至 15ml 离心管中,于 2500rpm 离心 5min。
2、 弃上清,加入 4ml PBS 并用枪轻轻吹打洗涤,然后 2500rpm 离心 5min。弃上清后用 PBS 重复洗涤一次。
3、 用枪洗干上清后,加 100μl 裂解液(含 PMSF)裂解30min,裂解过程中 要经常弹一弹以使细胞充分裂解。
4、 将裂解液与培养瓶中裂解液混在一起 4℃、12000rpm 离心 5min,取上清分装 于 0.5ml 离心管中并置于-20℃保存。 蛋白含量的测定 【 BCA方法】
1,从-20℃取出1mg/ml BSA,室温融化后,备用。取出BCA试剂盒
2,BCA工作液配制:A:B=50:1配制适量的BCA工作液,混匀,室温24h内稳定。 3,蛋白标准品配制:终浓度0.5 mg/ml
蛋白标准品 10ul PBS或生理盐水 90ul 注意:蛋白样品在什么溶液中,标准品也宜用什么溶液 4,样品和标准品均设置复孔 对照组
实验组
5,各孔加入200ul BCA工作液
6,37°孵育30min,测定A562,求平均值,绘制标准曲线。 注意:加样时注意避免气泡产生
【考马斯亮蓝方法】 (1) 制作标准曲线
1、 从-20℃取出 1mg/ml BSA,室温融化后,备用。
2、 2、 取 18 个 1.5ml 离心管,3 个一组,分别标记为 0μg,2.5μg,5.0μg ,10.0μg ,20.0μg ,40.0μg。 3、 按下表在各管中加入各种试剂。
4、 混匀后,室温放置 2min。在生物分光光度计(Bio-Photometer,Eppentoff) 上比色分析。、
(2) 检测样品蛋白含量
1、 取足量的 1.5ml 离心管,每管加入4℃储存的考马斯亮蓝溶液 1ml。室温放置30min 后即可用于测蛋白。
2、 取一管考马斯亮蓝加 0.15mol/L NaCl 溶液 100μl,混匀放置 2 分钟可做为空白样品,将空白倒入比色杯中在做好标准曲线的程序下按 blank 测空白样品。 3、 弃空白样品,用无水乙醇清洗比色杯 2 次(每次 0.5ml),再用无菌水洗一次。 4、 取一管考马斯亮蓝加 95μl 0.15mol/L NaCl NaCl 溶液和 5μl 待测蛋白样品,混 匀后静置 2min,倒入扣干的比色杯中按 sample 键测样品。
注意:每测一个样品都要将比色杯用无水乙醇洗 2 次,无菌水洗一次。可同时混合好多个样品再一起测,这样对测定大量的蛋白样品可节省很多时间。测得的结果是 5μl 样品含的蛋白量。
(二),SDS-PAGE 电泳
(1) 清洗玻璃板:
一只手扣紧玻璃板,另一只手蘸点洗衣粉轻轻擦洗。两面都擦洗过后用自来水冲,再用DDH2O冲洗干净后立在筐里晾干。 DDH2O:双蒸水 MQ:超纯水
(2) 灌胶与上样
1、 玻璃板对齐后放入夹中卡紧。然后垂直卡在架子上准备灌胶。
(操作时要使两玻璃对齐,以免漏胶。可用1%琼脂糖在垂直电泳槽的左,右和下部封边)
2、 按前面方法配 10%分离胶,加入 TEMED 后立即摇匀即可灌胶。灌胶时, 可用 10ml 枪吸取 5ml 胶沿玻璃放出,待胶面升到绿带中间线高度时即可。然后胶上加一层水,液封后的胶凝的更快。(灌胶时开始可快一些,胶面快到所需高度时要放慢速度。操作时胶一定要沿玻璃板流下,这样胶中才不会有气泡。加水液封时要很慢,否则胶会被冲变型;胶从一侧加,水从上部均匀加入;胶浓度<10%,可用0.1%SDS封,浓度>10%,用1ml水或异丁醇或异戊醇封,也可以用0.1%SDS封,封后,切记勿动) 3、 当水和胶之间有一条折射线时,说明胶已凝了。再等 3min 使胶充分凝固就可倒去胶上层水并用吸水纸将水吸干。若封胶用的醇应用水冲洗,然后加少量的0.1%SDS,目的是通过减少张力来清除残留水滴,片刻倒出SDS,后倒置玻璃板片刻控净。
4、等待凝胶的过程,,计算含 50-100μg 蛋白的溶液体积即为上样量。取出上样样品至 0.5ml 离心管中,加入 5×SDS 上样缓冲液至终浓度为 1×。(上样总体积一般不超过 15μl,加样孔的最大限度可加 20μl 样品。)上样前要将样品于沸水中煮 5min 使蛋白变性,亦可用PCR仪进行变性。然后放入-20℃保存。
5、按前面方法配 4%的浓缩胶,加入 TEMED 后立即摇匀即可灌胶。将剩余空 间灌满浓缩胶然后将梳子插入浓缩胶中。灌胶时也要使胶沿玻璃板流下以免胶中有气泡产生。插梳子时要使梳子保持水平。由于胶凝固时体积会收缩减小,从而使加样孔的上样体积减小,所以在浓缩胶凝固的过程中要经常在两边补胶。待到浓缩胶凝固后,两手分别捏住梳子的两边竖直向上轻轻将其拔出。
6 用水冲洗一下浓缩胶,将其放入电泳槽中。(小玻璃板面向内,大玻璃板面向外。若只跑一块胶,那槽另一边要垫一块塑料板且有字的一面面向外)
7、 加足够的电泳液后开始准备上样。(电泳液一定要超过玻璃板的上缘,否则电泳期间会出现电流过低的现象,影响电泳效果)用微量进样器贴壁吸取样品,将样品吸出不要吸进气泡。将加样器针头插至加样孔中缓慢加入样品。(加样太快可使样品冲出加样孔,若有气泡也可能使样品溢出。加入下一个样品时,进样器需在外槽电泳缓冲液中洗涤3 次,以免交叉污染。 上样(最外两电泳通道易跑歪,尽量不用) Marker: 6ul(Marker加在最边上的加样孔中) 样品: 20-30ul
实验所用的Marker电泳后,出现10条带;10,15,25,35,40,55,70,100,130,170KDa (3) 电泳:电压100V,电流300mA,功率50W,时间1.5h,电泳15min后可调节电压至130V,电泳至溴酚兰刚跑出即可终止电泳,进行转膜。(一定要让目的条带跑过分离胶的1/3;一般分离胶每胶20mA,浓缩胶每胶15mA,或恒压100V或120V;分离胶100V,浓缩胶75V )
电泳一般采用上层胶采用低电压恒压电泳,当溴酚蓝进入下层胶时使用高电压恒压电泳,对于Bio-Rad标准电泳装置和类似电泳装置,低电压设置在80-100V,高电压在120V左右。SDS-PAGF可以采用普通的电泳仪就满足了,为了方便,SDS-PAGF采用全过程恒压,通常电压设置在100V,时间设置在90-120min,设置时间应避免电泳过头 电泳结束前20min,配制转膜缓冲液
(三),转膜
(1),要先将玻璃板撬掉才可剥胶,撬的时候动作要轻,要在两个边上轻轻的反复撬。撬一会儿玻璃板便开始松动,直到撬去玻板。(撬时一定要小心,玻板很易裂。) 除去小玻璃板后,将浓缩胶轻轻刮去(浓缩胶影响操作),要避免把分离胶刮破。 其次,把胶放于蒸馏水中5min,重复3次(一直放于摇床摇动)最后,将胶浸于转移缓冲液中平衡 10 min(注意:如检测小分子蛋白,可省略此步,因小分子蛋白容易扩散出胶)摇床?。 (2),依据胶的大小剪取转移膜1片和滤纸 6 片,以及转膜用的夹子、两块海绵垫、一直玻璃棒或试管,放入转移缓冲液中平衡 10 min。如用 PVDF 膜需用纯甲醇浸泡饱和5min以上后转入TBS
液中平衡。(用镊子捏住膜的一边轻轻置于有超纯水的平皿里,要使膜浮于水上,只有下层才与水接触。这样由于毛细管作用可使整个膜浸湿。若膜沉入水里,膜与水之间形成一层空气膜,这样会阻止膜吸水)
注意:
a,切滤纸和膜时一定要戴手套,因为手上的蛋白会污染膜。 b,滤纸和NC 或PVDF膜应该与胶的大小一致。 c,放于缓冲液平衡时需摇床摇动吗
将切好的硝酸纤维素膜(左下角剪去一角,做标记)置于水上浸 2 h 才可使用。 (3) 将夹子打开使黑的一面保持水平。在上面垫一张海绵垫,用玻棒或试管来回擀几遍以擀走里面的气泡。(一手擀另一手要压住垫子使其不能随便移动。)在垫子上垫三层滤纸(可三张纸先叠在一起在垫于垫子上),一手固定滤纸一手用玻棒或试管擀去其中的气泡。
(4)将分离胶盖于滤纸上,用手调整使其与滤纸对齐,轻轻用玻棒擀去气泡。 将膜盖于胶上,要盖满整个胶(最好一次性成功,膜和胶接触后不可再移动)并除气泡。在膜上盖3张滤纸并除去气泡。最后盖上另一个海绵垫,擀几下就可合起
夹
Western
一、仪器与试剂及耗材
1)仪器:
2)试剂:
3)杂品与耗材:
二、试剂配制:
1、母液
1)1.0mol/L Tris-HCl
Tris (MW121.14) 30.29g
超纯水 200ml
溶解后,用浓HCl调 pH 至所需点(见下所示),最后用蒸馏水定容至 250ml,高温灭菌后室温下保存。
PH HCl
7.4 约 17ml
7.5 约 16ml(所需18.5)
7.6 约 15ml
8.0 约 10ml
2)1.5mol/L Tris-HCl(pH 8.8)(实际:用玻璃瓶配制50ml或100ml,其他材质会被盐酸腐蚀。)
Tris (MW121.14) 9.086g 18.172g 36.344g 45.43g
超纯水至 40ml 80ml 160ml 200ml
加浓盐酸量 1.4ml 2.8ml 5.6ml 7ml
定容至 50ml 100ml 200ml 250ml
溶解后,用浓盐酸调 pH 至 8.8,最后用超纯水分别定容至50ml、100ml、200ml 、 250ml,高温灭菌后室温下保存。
3)0.5mol/L Tris-HCl(pH 6.8)(实际:用玻璃瓶配制50ml或100ml,其他材质会被盐酸腐蚀。)
Tris (MW121.14) 3.028g 6.056g 12.112g
超纯水至 40ml 80ml 15.14g 160ml 200ml
加浓盐酸量 1.8ml 3.6ml 7.2ml 9.0ml
定容至 50ml 100ml 200ml 250ml
溶解后,用浓盐酸调 pH 至 6.8,最后用超纯水分别定容至50ml、100ml、 200ml、250ml,高温灭菌后室温下保存。
4)1.74mg/ml(10mmol)PMSF(实际:一次性离心管配制5或10ml)
PMSF 0.0174g 0.0435g 0.087g 0.174g
异丙醇 10ml 25ml 50ml 100ml 溶解后,高压灭菌,-20℃保存。
5)10%SDS(实际:一次性离心管配制5或10ml)
SDS 0.5g 1g 2g 10g
蒸馏水至 5ml 10ml 20ml 100ml
50℃水浴下溶解,室温保存。如在长期保存中出现沉淀,水浴溶化后,仍可使用。
6)10%过硫酸胺(AP)(实际:一次性离心管配制0.1或0.5ml)
过硫酸胺 0.01g 0.05
超纯水至 0.1ml 0.5ml
溶解后,4℃保存,保存时间为 1 周。最好现用现配
7)40%Acr/Bic(37.5:1)(实际:一次性离心管配制50ml。)
丙稀酰胺(Acr) 18.75g
甲叉双丙稀酰胺(Bic) 0.5g
超纯水至 50ml
37℃下溶解后,4℃保存。使用时恢复至室温且无沉淀。
8)20%Tween20(实际:一次性离心管配制10ml)
Tween20 2ml 5ml 10ml
蒸馏水至 10ml 25ml 50ml
混匀后 4℃保存。
9)0.2mol/L NaH2PO4
NaH2PO4(MW 119.98) 12g
蒸馏水至 500ml
溶解后,高压灭菌,室温保存。
10)0.2mol/L Na2HPO4
Na2HPO4 。12H2O(MW 358.14) 71.6g
蒸馏水至 1000ml
溶解后,高压灭菌,室温保存。
0.1g 1.0ml 37.5g 1g 100ml 20ml 100ml
2、使用液
1)单去污剂裂解液(50mmol/L TrisHCl pH8.0,150mmol/L NaCl,1%TritonX-100,
100μg/ml PMSF):
1mol/L TrisHCl(pH8.0) 2.5ml
NaCl 0.438g
TritonX-100 0.5ml
蒸馏水至 50ml
混匀后, 4℃保存。使用时,加入 PMSF 至终浓度为 100μg/ml(0.87ml 裂解液加入 1.74mg/ml PMSF 50μl)。
2)0.01mol/L PBS( pH 7.2-7.4)
0.2 mol/L NaH2PO4 19ml
0.2 mol/L Na2HPO4 81ml
NaCl 17g
蒸馏水至 2000ml
3)G250 考马斯亮蓝溶液(测蛋白含量专用)
考马斯亮蓝 G250: 100mg
95%乙醇 50ml
磷酸 100ml
蒸馏水至 1000ml
配制时,先用乙醇溶解考马斯亮蓝染料,再加入磷酸和水,混匀后,用滤纸过滤,4℃保存。
4)0.15 mol/L NaCl
NaCl(MW58.44) 0.877g
蒸馏水至 100 ml
高温灭菌后,室温保存。
5)100mg/ml 牛血清白蛋白(BSA)
BSA 0.1g
0.15 mol/L NaCl 1ml
溶解后,-20℃保存。制作蛋白标准曲线时,用 0.15 mol/L NaCl 进行 100 倍稀释成 1mg/ml,-20℃保存。
6)12%、10%分离胶; 4%、5%浓缩胶
12%分离胶 10%分离胶 4%浓缩胶 5%浓缩胶
(两块胶,10ml)(两块胶,10ml)(两块胶5ml)(两,5ml)
超纯水 3.2ml 4.85ml 3.16ml 3.5ml 40%Acr/Bic(37.5: 4.2ml 2.5ml 0.5ml 0.825ml
1.5 mol/L TrisHCl(pH8.8) 2.5ml 2.5ml - -
1.0mol/LTrisHCl(pH6.8) - - - 0.625ml 0.5 mol/L TrisHCl(pH6.8) - - 1.26ml - 10%SDS 100ul 100μl 50μl 50ul 10%AP(过硫酸胺) 55ul 50μl 25μl 50ul TEMED 5ul 5μl 5μl 10ul 加 TEMED 后,立即混匀即可灌胶。
(为了加快凝胶,可以将AP和TEMED量加大,一般加至原来的2-3倍;分离胶及浓缩胶均可事先配好(除AP及TEMED外),过滤后作为储存液避光存放于4度,可至少存放1个月,临用前取出室温平衡(否则凝胶过程产生的热量会使低温时溶解于储存液中的气体析出而导致气泡,有条件者可真空抽吸3分钟),加入
10%AP(0.7~0.8:100,分离 胶浓度越高AP浓度越低,15%的分离胶可用到0.5:100)及TEMED(分离胶用0.4:1000,15%的可用到0.3:1000,浓缩胶用0.8:1000)即可,如室温较低可升高10%AP及TEMED浓度到《分子克隆》建议浓度。) 注:TEMED避光保存
7)还原型 SDS(5X)上样缓冲液(0.25mol/L TrisHCl pH6.8,0.5mol/L 二硫叔糖
醇,10% SDS,0.5%溴酚蓝,50%甘油)
0.5 mol/L TrisHCl(pH6.8) 2.5ml
二硫叔糖醇(DTT,MW154.5) 0.39g
SDS 0.5g
溴酚蓝 0.025
甘油 2.5ml
混匀后,分装于 1.5ml 离心管中,4℃保存。
8)电泳液缓冲液 1X
(25mmol/L Tris,0.25mol/L 甘氨酸,0.1% SDS)
Tris(MW121.14) 3.03g 甘氨酸(MW75.07) 18.77g SDS 1g 蒸馏水至 1000ml 溶解后室温保存,次溶液可重复使用 3~5 次。
注意:
1、 电泳液可以回收重复利用,一般将回收的电泳液加入垂直电泳槽的下半部分,
上半部分最好用新鲜的电泳缓冲液
2、可以配成10X的电泳缓冲液保存,稀释10倍使用
9)半干转移缓冲液1X
(48mmol/L Tris,39mmol/L 甘氨酸,0.037% SDS,20%甲醇)
甘氨酸(MW75.07) 2.9g Tris(MW121.14) 5.8g SDS 0.37g 甲醇 200ml 蒸馏水至 1000ml
溶解后室温保存,次溶液可重复使用 3~5 次。
湿转电转液1X:
甘氨酸(MW75.07) 11.25g Tris(MW121.14) 2.375g SDS 0.375g 甲醇 200ml 蒸馏水至 1000ml
溶解后4℃保存,次溶液可重复使用3~5次 注意:
1、次溶液最好保存在4℃冰箱,最多使用5次左右,不然影响转移效率。 2、先用蒸馏水溶解甘氨酸、Tris和SDS,然后再加入甲醇,最后补足液体,
如果先加甲醇,溶解甘氨酸、Tris和SDS等比较困难。 3、可以配成2X转移液进行保存,稀释后使用
10)10X 丽春红染液
丽春红 S 2g 三氯乙酸 30g 磺基水杨酸 30g 蒸馏水至 100ml 使用时将其稀释 10 倍。
11)TBS 缓冲液(100mmol/ L TrisHCl pH7.5,150mmol/L NaCl)
1 mol/ LTrisHCl(pH7.5) 10ml NaCl 8.8g 蒸馏水至 1000ml
12)TBST 缓冲液(含 0.05%Tween20 的 TBS 缓冲液)
20%Tween20 1.65ml TBS 700ml 混匀后即可使用,最好现用现配。
13)封闭液(含 5%脱脂奶粉的 TBST 缓冲液)
脱脂奶粉(国产,安怡牌) 5g TBST 100ml
溶解后 4℃保存。使用时,恢复室温,用量以盖过膜面即可,一次性使用。
14)洗脱抗体缓冲液(100mmol/L 2-Mercaptoethanol,2%SDS,62.5m mol/L TrisHCl
pH6.8)
14.4 mol/L 2- Mercaptoethanol(β-巯基乙醇 700μl(通风厨里加)SDS 2g 0.5mol/L TrisHCl(pH6.8) 12.5ml 超纯水至 100ml 配制时,在通风厨内进行。4℃保存。可重复使用 1 次。
15)显影液(5X)
自来水(加热至 50℃) 375ml(以下药品加到温水中) 米吐尔 1.55g 亚硫酸钠(无水) 22.5g 碳酸钠(无水) 33.75g 溴化钾 20.95g 补水至 500ml
配制时,上述药品应逐一加入,待一种试剂溶解后,再加入后一种试剂。 4℃保存。使用时用自来水稀释至 1 倍。
16)定影液
自来水(50~60℃) 700ml (以下药品按顺序加入前者溶解后再加后者) 硫代硫酸钠 240g 亚硫酸钠(无水) 15g 冰乙酸 12.6ml 硼酸 7.5g
钾明矾 15g(水温冷至 30℃以下时再加入) 加水定容至 1000ml,室温保存
17)抗体
用 TBST 稀释至一定浓度使用,每张膜需0.5ml 18)化学发光试剂
购自北京中山公司,为 Santa Cruz 产品,分 A 和 B 两种试剂。 没钱可用碧云天的ECL
三、原理及简介 3.1,原理
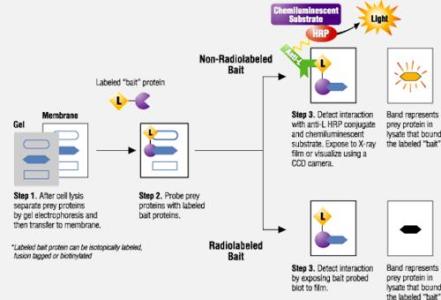
与Southern或Northern杂交方法类似,但Western Blot采用的是聚丙烯酰胺凝胶电泳,被检测物是蛋白质,“探针”是抗体,“显色”用标记的二抗。经过PAGE分离的蛋白质样品,转移到固相载体(例如硝酸纤维素薄膜)上,固相载体以非共价键形式吸附蛋白质,且能保持电泳分离的多肽类型及其生物学活性不变。以固相载体上的蛋白质或多肽作为抗原,与对应的抗体起免疫反应,再与酶或同位素标记的第二抗体起反应,经过底物显色或放射自显影以检测电泳分离的特异性目的基因表达的蛋白成分。该技术也广泛应用于检测蛋白水平的表达。
3.2 Western Blot 优点 1,高分辨率的电泳技术 2,特异敏感的抗原-抗体反应 3,1-5 ng 中等大小的靶蛋白
Western Blot 结合了凝胶电泳的高分辨率和固相免疫测定的特异敏感等多种优点,可检测到低至 1~5 ng(最低可到 10~100 pg)中等大小的靶蛋白。
3.3 Western Blot 应用 1,目的蛋白的表达特性分析
2,目的蛋白与其它蛋白的互作 3,目的蛋白的组织定位 4,目的蛋白的表达量分析
五 Western Blot基本流程解析
(一),蛋白的提取 1,
A,通过有机溶剂或水溶法制备总蛋白
水溶液提取法:针对水、稀盐、稀酸或碱溶液中的蛋白质。稀盐和缓冲系统的水溶液对蛋白质稳定性好、
溶解度大、是提取蛋白质最常用的溶剂 。 有机溶剂提取法:对于分子中非极性侧链较多的蛋白质可用有机溶剂提取,包括乙醇、丙酮和丁醇等。
B,通过层析或电洗脱法制备目的蛋白
2,裂解酶的选取
细胞及哺乳动物的组织:目前提取与纯化蛋白质最经典的方法是RIPA强、中、弱系列的裂解液及NP-40裂解液、SDS裂解液、哺乳动物蛋白抽提试剂,组织蛋白抽提试剂。这几种裂解液裂解原理相似,但作用效果不同,实验者需要根据自身实验需求来选取合适的裂解方式,例如RIPA(强)裂解液与SDS裂解液是作用效果很强的裂解液,可以使样本充分裂解,获取足够多的蛋白质,使蛋白质充分变性不具有蛋白质活性且较多的基因组等也伴随着提取出来了。RIPA中、弱裂解液及NP-40裂解液、哺乳动物蛋白抽提试剂、组织蛋白抽提试剂则较之温和,其缺点就是样本可能裂解不充分,核、膜蛋白可能提取不完全。具体的选择可以通过下附表进行选择。
细菌样本:细菌蛋白抽提试剂、包涵体蛋白溶解液。 酵母样本:酵母蛋白抽提试剂。 植物样本:植物蛋白抽提试剂。
细胞样本:细胞浆/核/膜蛋白抽提试剂盒。Western Blot实验主要是对蛋白质表达进行相对定量检测,如果有研究者需要对其进行细胞结构中蛋白质组分定位检测就需要用到此方法提取蛋白质,此方法是通过裂解液的裂解强度不同来分别提取同一细胞样本中不同结构的蛋白,这对于研究目的蛋白主要存在细胞于哪个结构中提供必要的技术。
在蛋白的提取过程中,因为实验样本如细胞、组织中本来就存在的各种蛋白酶在裂解过程中被液释放出来降解相应的蛋白质,所以在提取过程中不可避免的要使用到蛋白酶抑制剂,目前蛋白酶抑制剂的种类很多,最常用的是PMSF,最有效的是蛋白酶抑制剂混合物,其包含多种蛋白酶抑制剂,可以有效的防止蛋白在提取过程中蛋白的降解。
附表:
根据检测蛋白位置选择合适的裂解液
3,蛋白样品的定量
目前常用比色法测定样品蛋白的含量:Bradford 法(考马斯亮蓝法)、Lowry 法(Folin-酚试剂法) BCA、法等。但各有优缺点,大家可以根据具体情况选取。按相应蛋白质定量试剂盒操作说明操作,测定样品浓度。收集完蛋白样品后,为确保每个蛋白样品的上样量一致,需要测定每个蛋白样品的蛋白浓度。根据所使用的裂解液的不同,需要采用适当的蛋白浓度测定方法。因为不同的蛋白浓度测定方法对于一些去垢剂和还原剂等的兼容性差别很大,大家可以根据具体情况选取。
Bradford 法(考马斯亮蓝法):
考马斯亮蓝 G-250 有红、蓝两种不同颜色的形式,在一定浓度的乙醇和酸性条件下,可配成淡红色的溶液,与蛋白结合后形成蓝色化合物,该化合物在 595 nm 处有最大吸收值,化合物颜色深浅与蛋白的浓度高低成正比。 该法操作简便迅速,消耗样品量少,但不同蛋白质之间差异大,且标准曲线线性差。高浓度的 Tris、EDTA、尿素、甘油、蔗糖、丙酮、硫酸铵和去污剂对测定有干扰。缓冲液浓度过高时,改变测定液 pH 值会影响显色。
Lowry 法(Folin-酚试剂法):
福林-酚试剂(Folin-phenol reagent)的显色原理是:首先在碱性条件下蛋白质与铜作用生成蛋白质-铜复合物,该复合物随之将磷钼酸-磷钨酸还原成蓝色复合物。在一定条件下,蓝色强度与蛋白质量成正比,蛋白质浓度范围约在 25~250 μg/mL。
虽然该法是测定蛋白质浓度应用最广泛的方法之一,但其缺点也很明显,专一性较差,干扰物质多(如Tris 缓冲剂、蔗糖、硫酸铵、巯基化物、酚类、柠檬酸等),标准曲线的直线关系不特别严格。因此在其应用过程中有很多新的修正。 BCA 法(二喹啉甲酸法): 二喹啉甲酸(Bicinchoninic acid,BCA )法是Lowry测定法的一种改进方法,是近来广为应用的蛋白定量方法。其原理与Lowery法蛋白定量相似,即在碱性条件下蛋白质分子中的肽键能与Cu2+络合生成络合物,同时将Cu2+还原成Cu+。二喹啉甲酸及其钠盐是一种溶于水的化合物,在碱性条件下,可以和Cu+结合形成稳定的紫蓝色复合物,在 562 nm处有高的光吸收值,并且化合物颜色的深浅与蛋白质浓度成正比,据此可测定蛋白质浓度。
与 Lowery 法相比,BCA 蛋白测定方法操作更简单,试剂及其形成的颜色复合物稳定性更好,几乎没有干扰物质的影响,灵敏度高(微量检测可达到 0.5 μg/ml),应用更加灵活。与 Bradford 法相比,BCA 法的显著优点是不受去垢剂的影响。
BCA 法与 Lowry 法都容易受到蛋白质之间以及去污剂的干扰。Bradford 法敏感度最高,且与一系列干扰 Lowry,BCA 反应的还原剂(如 DTT,巯基乙醇)相容。但是对于去污剂依然是敏感的,其最主要的缺点
是不同的标准品会导致同一样品的结果差异较大,无可比性。
(二),SDA-PAGF 1,蛋白样品变性 1.1 SDS
阴离子去污剂、变性剂;
氨基酸侧链与 SDS 充分结合形成带负电荷的蛋白质-SDS 胶束;
蛋白质-SDS 胶束所带的负电荷大大超过了蛋白质分子原有的电荷量,消除了不同分子之间原有的电 荷差异;
与强还原剂一起使蛋白分子氢键、疏水键打开,使蛋白质分子线性化。
蛋白质-SDS 胶束的特点:
具有相同的形状,形状像一个长椭圆棒; 平均1g蛋白质结合14Gsds;
短轴对不同的蛋白质亚基-SDS胶束基本上是相同的; 长轴的长度则与亚基分子量的大小成正比。
1.2 SDS 聚丙烯酰胺凝胶电泳(SDS-PAGE) 1.2.1 SDS-PAGE 基本原理
1. SDS-PAGE 是在蛋白质样品中加入 SDS 和含有巯基乙醇的样品处理液,SDS 是一种很强的阴离子表面活性剂,它可以断开分子内和分子间的氢键,破坏蛋白质分子的二级和三级结构。
2. 强还原剂巯基乙醇(或二硫苏糖醇,DTT)可以断开二硫键破坏蛋白质的四级结构。使蛋白质分子 被解聚成肽链形成单链分子。解聚后的侧链与 SDS 充分结合形成带负电荷的蛋白质-SDS 复合物。 3. 蛋白质分子结合 SDS 阴离子后,所带负电荷的量远远超过了它原有的净电荷,从而消除了不同种蛋白质之间所带净电荷的差异。蛋白质的电泳迁移率主要决定于亚基的相对分子质量,而与其所带电荷的性质无关。
1.2.2 PAGE:聚丙烯酰胺凝胶
聚 丙 烯 酰 胺 凝 胶 (polyacrylamide gel) 是 由 单 体 丙 烯 酰 胺 ( acrylamide , 简 称 Acr ) 和 交 联 剂(crosslinker)N,N’-亚甲基双丙烯酰胺(N,N’-methylenebisacylamide,简称 Bis)在催化剂(过硫酸胺或核黄素 AP)和加速剂(四甲基乙二胺 TEMED)作用下聚合交联而成的三维网状结构的凝胶。化学惰性强,具有一定的机械强度和透明度。是良好的电泳介质。 聚丙烯酰胺凝胶聚合机理是通过提供氧游离基(free radicals)的催化,使体系发生氧化还原作用(catalyst-redox systems)来完成的。催化体系主要有化学催化(AP-TEMED)和光化学催化(核黄素-TMTED)体系。
1.2.3 聚丙烯酰胺凝胶分类
PAGE 分为连续系统和不连续系统两大类。连续系统电泳体系中缓冲液 pH 值与凝胶中的相同。带电颗粒在电场作用下,主要靠电荷和分子筛效应。不连续系统中带电颗粒在电场中泳动不仅有电荷效应、分子筛效应,还具有浓缩效应,因而其分离条带清晰度及分辨率均较前者佳。 不连续电泳
不连续系统的浓缩效应:
凝胶层的不连续性:浓缩胶的孔径大,分离胶的孔径小。在电场的作用下,蛋白质颗粒在大孔胶中遇 到的阻力小,移动快。而在小孔胶中遇到的阻力大,移动慢。因此,在两层凝胶的交界处,由于凝胶 孔径的不连续性使样品迁移受阻而压缩成很窄的区带。
缓冲液离子成分和pH的不连续性:HCl易解离出Cl-,它在电场中迁移率大,走在最前面,故称为快离 子或前导离子。电极缓冲液中的甘氨酸在pH6.8 的缓冲液中解离度很小,仅为 0.1-1%,因而在电场 中迁移率很小,称为慢离子或尾随离子。蛋白质均带负电荷,在电场中均移向正极,其有效迁移率介 于快慢离 子之间,于是蛋白质就在快慢离子间形成的界面处,被浓缩成极窄的区带。当进入pH8.8 的分离胶时,甘氨酸解离度增加,其有效迁移率超过蛋白质,因此氯离子和甘氨酸离子沿着离子界面 继续前进。蛋白质分子由于分子量大,被留在后面,然后分离成多个区带。
(三),转膜(Trarsmembran)
1,转膜的定义
将电泳后分离的蛋白质从凝胶中转移到固相载体(例如 NC 膜)上,通常有两种方法:毛细管印迹法和电泳印迹法。常用的电泳转移方法有湿转和半干转。两者的原理完全相同,只是用于固定胶/
膜叠层和施加电场的机械装置不同。前者操作容易,转移效率高;而后者适用于大胶的蛋
白转移,所用缓冲液少。
2,转移膜的选择
2.1 杂交膜的选择是决定 Western blot 成败的重要环节。应根据杂交方案、被转移蛋白的特性以
及分子大小等因素,选择合适材质、孔径和规格的杂交膜。
用于 Western blot 的转移膜主要有两种:硝 酸 纤 维 素 ( Nitrocellulose, NC ) 膜 和 聚 偏 二 氟 乙 烯(Polyvinylidene Fluoride, PVDF)膜,此外也有用尼龙膜、DEAE 纤维素膜做蛋白印迹。尼龙膜和 NC 膜的特点相似,主要用于核酸杂交。
2.2 NC 膜是蛋白印迹实验的标准固相支持物,在低离子转移缓冲液的环境下,大多数带负电荷的蛋
白质会与膜发生疏水作用而高亲和力的结合在一起,但在非离子型的去污剂作用下,结合的蛋白还可以被洗脱下来。根据被转移的蛋白分子量大小,选择不同孔径的 NC 膜。因为随着膜孔径的不断减小,膜对低分子量蛋白的结合就越牢固。通常用 0.45 μm 和 0.2 μm 两种规格的 NC 膜。大于 20 kD 的蛋白可用 0.45 μm 的膜小于 20 kD 的蛋白就要用 0.2 μm 的膜了,如用 0.45 μm 的膜就会发生“Blowthrough”的现象。
PVDF 膜灵敏度、分辨率和蛋白亲和力比常规的膜要高,非常适合于低分子量蛋白的检测。但 PVDF 膜在使用之前必需用纯甲醇浸泡饱和 1-5 秒钟。
2.3 硝酸纤维素(nitrocellulose, NC)膜:NC 与蛋白质靠疏水作用结合,无需预先活化,对蛋
白质的活性影响小;非特异性本底显色浅;价格低廉,使用方便。但结合在 NC 上的小分子蛋白质在洗涤时易丢失; NC韧性较差,易损坏。
聚偏二氟乙烯(Polyvinylidene fluoride, PVDF)膜:与蛋白质亲和力高,用前需在甲醇中浸泡,以活化膜上的正电基团,使其更容易与带负电荷蛋白结合。
2.4 膜的选择主要根据:
a,膜与目的蛋白分子的结合能力(也就是单位面积的膜能结合蛋白的载量),以及膜的孔径
(也就是拦截蛋白的大小);
b,不影响后续的显色检测(也就是适和用于所选的显色方法,信噪比好);
c,如果后继实验有其他要求,比如要做蛋白测序或者质谱分析,还要根据不同目的来挑选不同的转移膜。
几种膜的比较
(四),封闭(Blocking)
1, 杂交膜上有很多非特异性的蛋白质结合位点,为防止这些位点与抗体结合引起非特异的染色和背景,般用惰性蛋白质或非离子去污剂封闭膜上的未结合位点来降低抗体的非特异性结合。封闭剂应该封闭所有未结合位点而不替换膜上的靶蛋白、不结合靶蛋白的表位,也不与抗体或检测试剂有交叉反应。最常见的封闭剂是 BSA、脱脂奶粉、酪蛋白、明胶和 Tween-20(0.05 - 0.1%)稀溶液 PBST 或者 TBST。
5%脱脂奶粉或 BSA(室温孵育 1 h)
Western Blot 膜封闭液(生物试剂公司)
Tween-20 的作用:Tween-20 是一种非离子型去污剂,有复性抗原的作用,可提高特异性的识别能力。在做 westen blot 时,用惰性蛋白质或非离子去污剂封闭膜上的未结合位点可以降低抗体的非特异性结合。Tween 这种非离子型去污剂在乳化蛋白时,不破坏蛋白的结构,可减少对蛋白质之间原 有的相互作用的破坏。离子型去污剂如 SDS 则破坏蛋白的结构。
2,封闭剂选择的特殊情况:
a,脱脂奶粉不能与生物素化或伴刀豆蛋白标记的抗体一起使用,因为脱脂奶粉含有糖蛋白和生物素
b,如果封闭剂中含磷酸酶,用磷酸化特异性抗体分析磷酸化蛋白受到影响,因为磷酸酶与印记膜上的磷酸化蛋白接触可使之去磷酸化。
C,检测磷酸化抗体时,不能使用酪蛋白/脱脂奶粉作为封闭剂。
3,一般封闭条件为
室温或者 37℃缓慢摇荡 1~2 h,特殊情况也可 4℃过夜。根据结果情况调整封闭试剂的浓度和类型。比如有时 BSA 比脱脂奶粉更有利于降低非特异性背景,而有些抗体则在脱脂奶粉封闭的情况下才能得到清晰地背景。
(五),一抗、二抗孵育(Antibody incubation)
1,一抗孵育(Primary antibody incubation)
参考一抗的说明书,按照适当比例用封闭液稀释一抗。
吸尽封闭液后,立即加入稀释好的一抗,室温或 4℃在摇床上缓慢摇动孵育 1~2 小时。如果一抗孵育一小时效果不佳,可以在 4℃缓慢摇动孵育过夜。(注意:抗体浓度过高会导致背景较深,过低会导致和抗原结合不充分,出现假阴性 )。回收一抗。然后加入 Western 洗涤液,在摇床上缓慢摇动洗涤 5-10 分钟。吸尽洗涤液后,再加入洗涤液,洗涤 5-10 分钟。共洗涤 3 次。如果结果背景较高可以适当延长洗涤时间并增加洗涤次数。
2,二抗孵育(Secondary antibody inucubation)
参考二的说明书,按照适当比例用封闭液稀释标记的二抗
吸尽封闭液后,立即加入稀释好的二抗,室温4℃在摇床上缓慢摇动孵育 1~2 小时。回收二抗 如果一抗孵育一小时效果不佳,可以在 4℃缓慢摇动孵育过夜。然后加入 Western 洗涤液,在摇床上缓慢摇动洗涤 5-10 分钟。吸尽洗涤液后,再加入洗涤液,洗涤 5-10 分钟。共洗涤 3 次。如果结果背景较高可以适当延长洗涤时间并增加洗涤次数。
前 2 步洗涤是为了洗去一抗和二抗的非特异性结合,洗涤的效果直接影响结果背景的深浅,所以洗涤一定要干净。洗涤液使用 TBST 或者 PBST,其中的 Tween20 有助于去除非特异性的结合,减少背景。同时短时间多次的洗膜(如 5-6 次,每次 3 分钟)比长时间少次数的洗膜更有效。 第 3 步 TBS 洗涤是为了洗去膜上的 Tween-20,因为它可以阻碍底物的沉积。
注意事项
1. 抗体保存注意事项:
收到抗体后按要求离心后再打开管盖进行分装和保存!
对于大部分抗体,比较合适的保存方式是分装后保存在-20℃ 。分装的量以一次实验用完为好,最少不能少于 10 ul 每份。因为分装体积越小,抗体的浓度越可能会受到蒸发以及管壁吸附的影响 复融后的分装抗体如果一次用不完,将剩余母液保存在 40℃,避免再冻起来!
绝对避免将抗体保存在自动除霜冰箱中。
大部分抗体收到后 40℃短暂保存 1-2 周对抗体活性是没有影响的。
长期保存,加入叠氮纳,防止细菌污染。
2. 一抗/二抗使用问题:
一抗、二抗的浓度一般要参照抗体说明书选择最适当的比例,一抗二抗的选择直接影响实验结果以及背景的深浅。要严格保证反应时间,洗膜要注意尽可能地将一抗二抗洗净,有利于降低背景;还要注意一抗二抗的匹配。
3. 天然和非还原样品问题:
有些抗体只能识别的表位是非连续氨基酸,这些氨基酸虽然在蛋白质一级结构中彼此不连续,但是在空间结构中则是靠在一起的。这些抗体就只能识别靶蛋白的空间结构状态(抗体说明书中会有特殊说明)。对于缓冲液和凝胶中就不能含有 SDS,并且不能够对蛋白进行加热变性。
有些抗体只能识别蛋白的非还原状态如氧化状态,对此类样品,就需要将缓冲液和凝胶中的还原剂 DTT和 beta-巯基乙醇去除。
(六)蛋白检测(Detection of proteins)——显影
酶促反应比同位素安全且快速,已经成为 Western Blot 的主流检测方法。酶促反应可以搭配不同的底物从而实现不同的显色方法:化学发光和底物显色,前者灵敏度很高,已经达到皮克级别,甚至还有飞克级别的,灵敏度超过了同位素;而后者由于直接显色而操作简便且成本低。
大家最为熟悉熟悉当数辣根过氧化物酶 HRP(Horseradish Peroxidase)和碱性磷酸酶 AP
(AlkalinePhosphatase) 此外还有比较少见的葡萄糖氧化酶,(Glucose Oxidase) β 半乳糖苷酶和(β-Galactosidase)。
在酶连抗体中使用的酶通常是碱性磷酸酶或辣根过氧化物酶。碱性磷酸酶可以将无色的底物 5-溴-4-氯吲哚磷酸盐(BCIP)转化为蓝色的产物;而辣根过氧化物酶可以以H2O2为底物,将 3-氨基-9-乙基咔唑氧化成褐色产物或将 4-氯萘酚氧化成蓝色产物。另一种检测辣根过氧化物酶的方法是用增强化学发光法,辣根过氧化物酶在H2O2存在下,氧化化学发光物质鲁米诺(luminol,氨基苯二酰一肼)并发光,在化学增强剂存在下光强度可以增大 1000 倍,通过将印迹放在照相底片上感光就可以检测辣根过氧化物酶的存在。
① HRP 标记二抗用 DAB 显色,按顺序依次将 DAB 三种剂各取 3 滴于 5 ml 蒸馏水中,避光混匀,将膜加入显色液中避光显色 5-15min 终止反应,对照 Marker 记录实验结果, NC 膜晾干扫描保存。将HRP的生色底物显色有 DAB、4-CN、CN/DAB、AEC 和 TMB 等。
HRP 化学发光反应底物主要有 Luminol、ECL 和 SuperSignal 系列。可直接从公司购买试剂盒,操作比较简单,原理如下(二抗用 HRP 标记):反应底物为过氧化物+鲁米诺,如遇到 HRP,即发光,可使胶片曝光,就可洗出条带。
② AP显色底物生成的产物主要是蓝紫色,成像性好容易拍照。 AP的底物显然更稳定,不像HRP那样需要 新 鲜 配 制 的 H2O2 一 类 的 不 稳 定 成 分 , 作 为 试 剂 盒 可 以 保 存 时 间 更 长 。 常 用 显 色 底 物 的 是 BCIP(5-Bromo-4-Chloro-3 ’ -Indolyphosphate p-Toluidine Salt) , NBT (Nitro-Blue Tetrazolium
Chloride)和INT。
③ 底物发光法:将两种显色底物 1:1 等体积混合后将其覆盖在膜表面使其均匀,用玻璃胶片把膜包起来, 马上在暗室中将 X 光片覆盖在膜的上面(时间根据光的亮度来衡量),显影、定影。(注意:发光法检测的时候曝光时间要恰到好处,过短会导致曝光不彻底,影响条带亮度,过长会导致荧光信号衰弱,也会使背景颜色加深)
除了酶连二抗作为指示剂,也可以使用其它指示剂,比如:荧光素异硫氰酸盐标记的二抗(可通过紫外灯产生荧光);生物素结合的二抗等等。
关于磷酸化蛋白检测的注意事项:
① 保持待测蛋白处于磷酸化状态,在标本提取液中加入足够的磷酸酶抑制剂(NaF,可以防止去磷酸化),并将标本时刻保持在冰浴中。
② 使用 5%的 BSA 作为封闭试剂,不能使用脱脂奶粉(因为脱脂奶粉中含有酪蛋白,会出现很高的背景)。
③ 请谨记蛋白的磷酸化是需要诱导的,当信号低或者无信号时有可能是磷酸化诱导不充分!确保使用阳性对照。
八,膜的重复利用(Membrane recovery)
8.1
a,膜解析的方法
. 通过加热和去污剂进行解吸,原理:组合使用去污剂和加热使抗体解离,适用于任何化学发光底物系统。
b,酸解吸,原理:使用低 pH 改变抗体结构从而使结合位点失活,从而将抗体与抗原分离,适用于任何化学发光底物系统。
两种方法都不能去除由显色检测系统产生的有色沉淀物(例如 BCIP、4-CN、DAB 和 TMB)。然而,仍可以用针对不同靶蛋白的特异性抗体分析印迹膜。
重要提示:在各次免疫检测之间不应使印迹膜干燥。印迹膜干燥将会使任何剩余抗体与膜永久性结合。
8.2通过加热和去污剂进行解吸:
仪器和溶液:
解吸液:100 mM 2-巯基乙醇,2%(w/v)SDS,62.5 mM Tris-HCl,pH6.7,磷酸盐缓冲液(PBS):10 mM 磷酸钠,pH7.2,0.9%(w/v)NaCl,浅托盘,大小足以将膜放入其中。
操作:
a. 在通风橱中,将印迹膜放入解吸液中在 50℃下振摇 30 分钟。
b. 将印迹膜放入缓冲液中振摇 10 分钟。用新缓冲液重复漂洗两次。
c. (可选)重复起始检测方法(忽略一抗步骤),确保已灭活抗体或已从膜上解吸抗体。 d. 将印迹膜放入缓冲液中振摇 10 分钟。
e. 继续下一轮检测的封闭步骤。
2. 酸解吸
仪器和溶液:
解吸液:25 mM 甘氨酸- HCl,pH2,1%(w/v)SDS,磷酸盐缓冲液(PBS):10 mM 磷酸钠,pH7.2,0.9%(w/v)NaCl,浅托盘,大小足以将膜放入其中。
操作:
a. 将印迹膜放入解吸液中振摇 30 分钟。
b. 将印迹膜放入缓冲液中振摇 10 分钟。用新缓冲液重复漂洗。
c. 继续下一轮检测的封闭步骤。
九 ,操作详细步骤
蛋白样品制备SDA-PAGF转膜封闭一抗二抗洗膜
蛋白检测-显色或显影
(一), 蛋白样品制备
(1) 单层贴壁细胞总蛋白的提取:
1.培养的细胞(定性):
⑴ 去培养液后用温的PBS冲洗2~3遍(冷的PBS有可能使细胞脱落)。
⑵ 对于6孔板来说每孔加200~300μl,60~80℃的1×loading buffer。 ⑶ 100℃,1min。
⑷ 用细胞刮刮下细胞后在EP管中煮沸10min,期间vortex 2~3次。
⑸ 用干净的针尖挑丝,如有团块则将团块弃掉,如果没有团块但有拉丝现象,则可以将EP管置于0℃后在14000~16000g离心2min,再次挑丝。若无团块也无丝状物但溶液有些粘稠,可通过使用1ml注射器反复抽吸来降低溶液粘滞度,便于上样。
⑹ 待样品恢复到室温后上样。
2.培养的细胞(定量):
⑴ 去培养液后用温的PBS冲洗2~3遍(冷的PBS有可能使细胞脱落)。
⑵ 加入适量的冰预冷的裂解液后置于冰上10~20min。
⑶ 用细胞刮刮下细胞,收集在EP管后超声(100~200w)3s,2次。 ⑷ 12000g离心,4℃,2min。
⑸ 取少量上清进行定量。
⑹ 将所有蛋白样品调至等浓度,充分混合沉淀后加loading buffer后直接上样最好,剩余溶液(溶于1×loading buffer)可以低温储存,-70℃一个月,-20℃一周,4℃ 1~2天,每次上样前98℃,3min。
1、 按 1ml 裂解液加 10μl PMSF(100mM)[0.87ml 裂解液加入 1.74mg/ml PMSF 50μl],摇匀置于冰上。(PMSF 要摇匀至无结晶时才可与裂解液混合。)
2、 倒掉培养液,并将瓶倒扣在吸水纸上使吸水纸吸干培养液(或将瓶直立放置一 会儿使残余培养液流到瓶底然后再用移液器将其吸走)。
3、去培养液后用温的PBS冲洗2~3遍(冷的PBS有可能使细胞脱落)。【培养瓶:每瓶细胞加 3ml 温的 PBS(0.01M pH7.2~7.3) 培养板:】。平放轻轻摇动 1min 洗涤细胞,然后弃去洗液。重复以上操作两次,共洗细胞三次以洗去培养液。将PBS 弃净后把培养瓶或板置于冰上。
4、 每瓶细胞加 400μl 含 PMSF 的裂解液或六孔培养板每孔150-200ul,12孔板50-60ul,于冰上裂解 30min,为使细胞充分裂解培养瓶要经常来回摇动。(可放置冰袋上,然后一块放在4℃冰箱)
5、 裂解完后,用干净的刮棒将细胞刮于培养瓶的一侧(动作要快,处理不同的样品之前清洗刮刀),然后用枪将细胞碎片和裂解液移至 1.5ml 离心管中后超声(100~200w)3s,2次。(整个操作尽量在冰上进行。)
6、 于 4℃下 12000rpm 离心 5min。(提前开离心机预冷)(分三层:上层为油脂,中层为蛋白,下层为沉淀)
7、 将离心后的上清分装转移倒 0.5min 的离心管中放于-20℃保存。 本人:
1,参考下面,直接在有培养液时用细胞刮刀刮起细胞,然后用枪头吹打,后转移到离心管,2500rp离心5min,用PBS清洗一次,2500rp离心5min弃上清后,用 PBS 重复洗涤一次。
2、 用枪吸干上清后,加 100μl 裂解液(含 PMSF)裂解30min,裂解过程中 要经常弹一弹以使细胞充分裂解。
4、 将裂解液与培养瓶中裂解液混在一起 4℃、12000rpm 离心 5min,取上清分装于 0.5ml 离心管中并置于-20℃保存。
子。整个操作在转移液中进行,要不断的擀去气泡。膜两边的滤纸不能相互接触,接触后会发生短路。注意膜在正极+侧,分离胶在负极-侧。标记正反面。(转移液含甲醇,操作时要戴手套,实验室要开门以使空气流通。)
(5) 将夹子放入转移槽槽中,要使夹的黑面对槽的黑面,夹的白面对槽的红面。电转移时会产热,放在冰盒内或在槽的一边放一块冰来降温。一般用200mA 1至2小时或恒压100V 1到2小时。一般用 60V 转移 2 h 或 40V 转移3 h。
(6)转膜后检测(此步可以省略)
转膜完毕后,立即把蛋白膜放置到预先准备好的 TBST(或 PBST)中漂洗 1-2 分钟,以洗去膜上的转膜液。转膜的效果可以观察所使用的预染蛋白质分子量标准,通常分子量最大的 1-2 条带较难全部转到膜上。转膜的效果也可以用丽春红染色液或硬度墨汁染色液对膜进行染色,以观察实际的转膜效果。也可以用考马斯亮蓝快速染色液对完成转膜的 SDS-PAGE 胶进行染色,以观察蛋白的残留情况。然后将膜晾干备用。
a. 丽春红染色:蛋白带出现后,于室温用去离子水漂洗硝酸纤维素滤膜,换水几次。 b. 印度墨汁染色:只用于放射性标记抗体或放射性标记 A 蛋白探针的 Western 印迹过程。
印度墨汁不能和化学发光物合用,若是使用呈色反应的酶检测法时,印度墨汁的黑色会使凝胶难于拍照。这时,可改用其他检测方法或换用丽春红 S。
注意:从转膜完毕后所有的步骤,一定要注意膜的保湿,避免膜的干燥,否则极易产生较高的背景。
转移方法的选择:
? 半干转
?
?
?
? 用滤纸吸buffer来做转移体系的转移方法; 适合转移小分子量的蛋白; 半干转的电流大小是按照面积来算的,时间是根据蛋白分子大小定的; 1.2-2MA/CM2 1h
? 湿转
?
?
?
?
将膜、胶、滤纸整个浸泡在buffer的Tank里转移的方法; 适合转移大分子量(100KD以上)蛋白; 湿转电流是恒定的,时间也是根据分子量而定; 300mA 1h
转膜的注意事项
1. 泡膜:转膜之前将海绵、胶、膜都用预冷的转移 buffer 浸泡 20min;
a. 凝胶若是没在预冷的转膜 buffer 中浸泡,就会在转膜过程中出现凝胶皱缩,导致出现转移条带变形。
b. PVDF 膜具有疏水性,需用甲醇浸泡。
2. 转膜顺序:阴极碳板+海绵+三滤+胶+膜+三滤+海绵+阳极碳板;
3. 转膜条件:0.35 A/250 V/1 h/40 min/冰浴中进行转膜(转膜过程产生大量的热,注意冷却);
(具体转膜时间要根据目的蛋白的大小而定;目的蛋白的分子量越大,需要的转膜时间越长,反之则短。)
4. 其它注意事项:
a. 避免直接用手碰杂交膜,使用镊子。因为手上的蛋白和油脂会影响转膜效率并会使膜脏掉。
b. 夹好膜和凝胶后,确定在凝胶/膜和滤纸之间没有气泡存在,否则会导致转膜不完全。
c. 保证膜和滤纸的大小和凝胶完全一样,过大和过小都会影响转膜效率。
d. 鸡来源的抗体与 PVDF 和尼龙膜有较强的结合能力,从而产生较高的背景,故如果选择鸡来源的抗体
(四),封闭
将膜用去离子水或TBS还是TBST 从下向上浸湿后,移至含5%脱脂奶粉的TBST(10mM Tris-HCl, pH7.5,150mM NaCl, 0.05%Tween-20)的封闭液的平皿中,室温下脱色摇床上摇动封闭 1h。
(五),一抗孵育
A, 用parafilm膜制作与膜大小相同的槽
B,从封闭液中取出膜,用滤纸吸去残留液后,用TBST清洗3×5min;
C,将一抗用 一抗稀释液或TBST 稀释至适当浓度(在 1.5ml 离心管中) D, 将膜蛋白面朝上放于槽中,加入抗体,室温下孵育 1~2h 后,4℃过 E, 次日,用 TBST 在室温下脱色摇床上洗3次,每次 10min;再用 TBS 洗
一次,10min。
(六),二抗孵育
(3) 同上方法准备二抗稀释液并与膜接触,室温下孵育 1~2h 后,用 TBST 在室温下脱色摇床上洗3次,每次 10min;再用 TBS 洗一次,10min,进行化学发光 反应。
注意:
1,一抗和二抗孵育使用杂交袋,并用封膜机将膜蛋白密封
(七),蛋白测定----化学发光,显影,定影
(1)按1:1的体积比配制化学发光液,混匀,置于保鲜膜上;1min 后,将膜蛋白面朝下与此混合液充分接触;1min 后,将膜移至另一保鲜膜上,去尽残液,包好,放入 X-光片夹中。
(2) 在暗室中,将 1×显影液和定影液分别到入塑料盘中;在红灯下取出 X-光片,用切纸刀剪裁适当大小(比膜的长和宽均需大 1cm);打开 X-光片夹,把 X-光片放在膜上,一旦放上,便不能移动,关上 X-光片夹,开始计时;根据信号 的强弱适当调整曝光时间,一般为 1min 或 5min,也可选择不同时间多次压片, 以达最佳效果;
曝光完成后,打开 X-光片夹,取出 X-光片,迅速浸入显影液中显影,待出现明显条带后,即刻终止显影。显影时间一般为 1~2min(20~25℃),温度过低时(低于 16℃)需适当延长显影时间;
显影结束后,马上把 X-光片浸入定影液中,定影时间一般为 5~10min,以胶片透明为止;用自来水冲去残留的定影液后,室温下晾干。
应注意的是:显影和定影需移动胶片时,尽量拿胶片一角,手指甲不要划伤胶片,否则会对结果产生影响。
(七) 凝胶图象分析
将胶片进行扫描或拍照,用凝胶图象处理系统分析目标带的分子量和净光密度值。
(八),结果分析
1,对照设计
成功进行Western Blot 检测,设置合适正确的对照是必不可少的,只要正确设置了这些对照,即可快速和准确的找到 Western Blot 的问题所在,并保证实验结果的准确性和特异性!
一般需要设置的对照如下:
阳性对照:明确表达检测蛋白的组织或细胞,用于检测一抗抗体的工作效率及一抗是否正常; 阴性对照:明确不表达检测蛋白组织或细胞,用于检测一抗抗体的特异性;
二抗对照:不加一抗,用于检测二抗的特异性;
内参对照:检测标本的质量和二抗系统;
空白对照:不加一抗和二抗;用于检测膜的性质和封闭的效果。
2,内参的选择
(一),简介
内参即内部参照,对于哺乳动物细胞表达来说一般是指由管家基因编码表达的蛋白,它们在各组织和细胞中的表达相对恒定,在检测蛋白的表达水平变化时常用它来做参照物。在 Western Blot 中使用内参其实就是在 WB 过程中另外用内参对应的抗体检测内参,这样在检测目的产物的同时可以检测内参的表达,由于内参在各组织和细胞中的表达相对恒定,借助检测每个样品内参的量就可以用于校正蛋白质定量、上样过程中存在的实验误差,保证实验结果的准确性。此外使用内参可以作为空白对照,检测蛋白转膜情况是否完全、整个 Western Blot 显色或者发光体系是否正常。
一般要选择一个在处理因素作用的条件下蛋白含量不会发生改变的蛋白作内参
(二),内参的选择
内参抗体种类很多,比如β-actin、β-tubunlin、GAPDH、Lamin B等,那么针对自己的实验,我们该如何选择呢?下面简单介绍下选择内参抗体应遵循的原则:
1,样本种属来源:
首先要考虑的就是实验样本来源于什么物种。
(1)、哺乳动物的组织或者细胞样本,通常选择β-actin、β-tubulin、GAPDH、Lamin B、Histone H3、Na,Katpase等。
(2)、植物来源实验样本,则可以选择plant actin、Rubisco等。
(3)、其他来源样本研究较少,所以就应该参照文献报导,选择合适的蛋白作为内参。 2,目的蛋白分子量:
选择内参抗体时,应该考虑目的蛋白分子量的大小。通常应该保证目的蛋白与内参蛋白分子量相差5KD以上。比如目的蛋白分子量为45KD,此时不适宜选择β-actin作为内参,可以考虑选择GAPDH或者β-tubulin作为内参。
3,目的蛋白表达部位:
就一般的蛋白检测来说,β-actin、β-Tubulin抗体等就可以了,而针对于核蛋白的定量,特别是样本蛋白就是核蛋白时,选择恰当的核蛋白内参则更能体现内部参照的价值。常用的核内参抗体有Lamin A、Lamin B、Histone H3,除此之外,其它常见的核蛋白内参还有PCNA、K70、K80等,在一些文献报道中,Erk2、TATA binding protein(TBP)以及c-Jun、c-Fos等都有使用。而对于膜蛋白检测,常用的内参抗体为Na,K ATPase。对于线粒体蛋白的检测,常用VDAC1和COX IV作为内参抗体。
以上几条原则只是针对通常情况,但是需要注意的问题是——内参的选择需要考虑实际的试验环境,比如某些细胞中,由于组织缺氧、糖尿病等因素会导致GAPDH的表达增高,不适合做内参。比如在涉及细胞增殖相关试验中,c-Jun由于自身表达变化就不适合做内参;而在凋亡实验时,TBP、Lamin等也不适合作为内参。因此设计实验方案的时候应该考虑这些因素并查询相应文献,在实验过程中也应该注意如果内参表达出现异常,应考虑这方面因素。
百度搜索“爱华网”,专业资料,生活学习,尽在爱华网