最近“冰桶挑战”在网上大热,这使得人们关注到一个神经内科的罕见病:肌萎缩性侧索硬化(amyotrpnic lateral sclerosis,ALS),这令作为一个从事神经内科医生来说感到十分的欣慰。自2006年至今,在过去8年的时间里,我们共接诊治疗了125位确诊为肌萎缩侧索硬化症的患者,这个疾病对于普通人来说很陌生,但却是一种致死率很高的疾病,目前尚没有有效的治疗方法。
ALS的流行概况
肌萎缩性侧索硬化(amyotrpnic lateral sclerosis,ALS),又称为卢伽雷氏症(Lou Cehrigs disease),俗称渐冻人症。1869年由让-马丁·沙可(Jean—Martin Charcot)首次确诊。它是一种不可逆的致死性运动神经元疾病。病变主要侵犯上运动神经元(大脑、脑干、脊髓),又影响到下运动神经元(颅神经核、脊髓前角细胞)及其支配的躯干、四肢和头面部肌肉的一种慢性进行性变性疾病。
让-马丁·沙可,19世纪法国神经学家、解剖病理学教授。他的工作大大推动了神经学和心理学领域的发展。他的绰号是“神经症领域的拿破仑”。图片来源:wikipedia.org
在美国ALS的报告发病率(每年新发病例)为2/10万-4/10万,患病率为4/10万-6/10万,中国尚无确切的流行病学资料。 ALS分为家族性和散发性两种类型。散发性以男性多见,男女之比约为1.5∶1-2∶1。中年后起病,多数患者为50-70岁,平均发病年龄为55岁,40岁以下发病也有报告,20-30岁发病约占5%。 家族性ALS约占5%-10%,多为常染色体显性遗传,男女发病率相等,发病年龄相对于散发性较早,平均约为49岁。
在我们接诊的125例病例,男女比例为1.6:1,多数为中年后起病,最小发病年龄32岁,最大发病年龄59岁,平均发病年龄为54岁,均为家族中首例发病患者。
自首发症状后,患者的平均生存期为3-5年,有些患者的生存期仅为数月,而有些患者生存期长达10余年,最后多死于呼吸肌麻痹导致的呼吸衰竭。而像霍金老爷子病程进展如此缓慢的十分罕见,可能这也算是他不幸中的万幸了。
目前关于该病的病因及发病机制有很多理论及假说,但具体病因和发病机制仍旧不清楚。
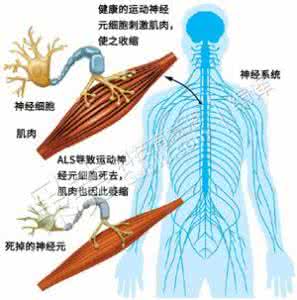
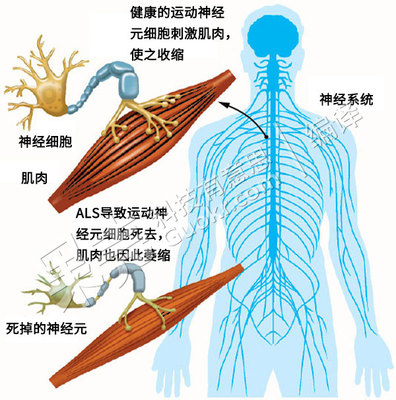
ALS是一种渐进性的神经退行性疾病,它影响大脑和脊髓中与运动相关的神经细胞,造成运动神经元死亡,令大脑无法控制肌肉运动,肌肉也会因缺乏运动而萎缩。图片来源:freep.com 编译:Ent
ALS的临床特点
起病隐匿,缓慢进展。
半数患者首发症状为肢体无力伴肌萎缩(5%)和肌束颤动(4%),上肢远端尤其突出。此时四肢腱反射减低,看不到病理征。随着病情的发展患者逐渐出现典型的上下运动神经元损害的体征,表现为广泛而严重的肌肉萎缩、肌张力增高、病理征表现为阳性。
60%的患者具有明显的上下运动神经元体征。而当下运动神经元变性达到一定程度时,肌肉广泛失神经,此时可无肌束颤动腱反射减低或消失,也无病理征。
约有10%的患者在整个病程中仅表现为进行性的肌肉萎缩而无上运动神 经元损害的体征。
约30%的患者以脑干的运动神经核受累起病,表现为吞咽困难、构音不清、呼吸困难、舌肌萎缩和纤颤,以后逐渐累及四肢和躯干,情绪不稳定(强哭强笑)是上运动神经元受累及假性延髓性麻痹的征象。
以脊髓侧索受累为首发症状的肌萎缩侧索硬化罕见。9%的患者可有痛性痉挛,多在受累的下肢近端出现,常见于疾病的早期。10%的患者有主观的肢体远端感觉异常或麻木除非合并其他周围神经病,ALS无客观的感觉体征。整个病程中膀胱和直肠功能保持良好,眼球运动通常不受损害。
单纯的ALS患者一般没有智力减退。ALS伴有其他神经系统变性疾病的症状和体征时,称ALS叠加综合征(ALS-plussyndrome)。该综合征主要发生在西太平洋地区、日本的关岛和北非等地区。合并的症状和体征包括锥体外系症状小脑变性、痴呆自主神经和感觉系统症状以及眼球运动异常。
神经肌肉电生理改变主要表现为广泛的神经源性损害。ALS的神经源性损害通常累及3个以上的区域。
对于ALS的诊断,初步诊断一般多依靠病史、体检和电生理检查。而临床确诊一般通过临床或神经电生理检查,证实确实存在上、下运动神经元同时受累的证据。另外还要与颈椎病、腰椎病、脊髓性肌萎缩等多种疾病相鉴别。
ALS的治疗
尽管ALS仍是一种无法治愈的疾病,但有许多方法可以改善患者的生活质量,应早期诊断,早期治疗,尽可能延长生存期。现阶段研究方向包括抗兴奋毒性药物、抗氧化和自由基清除剂、神经营养因子、基因治疗等方面,此外还要重视支持和对症治疗。
延缓病情发展的药物:
(1)抗兴奋毒性药物:目前美国FDA对此病唯一批准的治疗用药是抗兴奋性氨基酸毒性的药物利鲁唑(Riluzole),是一种谷氨酸的拮抗剂,可以减少谷氨酸引起的细胞激活毒性。无论是对延髓麻痹还是肢体瘫痪为首发症状的ALS患者都有一定疗效,可以延缓ALS的进展。1994年和1996年分别进行的两个大规模随机双盲安慰剂对照的临床研究,证实了该药虽不能治愈ALS和改善症状,但能够明确的延长生存时间和推迟气管切开的时间。目前该药仍在临床上广泛使用,但是疗效远远不能显著改善ALS患者的生存质量。当病程晚期患者已经使用有创呼吸机辅助呼吸时,不建议继续使用。(2)其他药物:在动物实验中,尽管有多个药物在ALS动物模型的治疗中显示出一定的疗效,如肌酸、大剂量维生素E、辅酶Q10、碳酸锂、胰岛素样生长因子-1、睫状神经营养因子、脑源性神经营养因子、胶质细胞源性神经营养因子、拉莫三嗪、头孢曲松钠,格拉默,二甲胺四环素,超氧化歧化酶基因SODl的反义化合物等,但在针对ALS患者的临床研究中均未能证实有效。
基因治疗:
最近发现ALS患者有基因突变,因此推测利用基因介导技术可以治疗突变基因的过度表达,可能具有一定的治疗作用。国内外大量研究者正在为证实此治疗理论不断研究。
免疫治疗:
近年来,随着免疫学及分子生物学技术的发展,免疫因素在AIS发病机制中的作用得到广泛重视。导致AIS的免疫机制复杂,其中关于抗钙通道抗体学说的研究较多。实验证明,应用电压依从性钙通道的阻滞剂匹莫齐特(Pimozidete)的ALS患者的病情进展指数比服用神经保护刺司来吉兰(selegiline)和维生素E的患者低,说明钙通道阻滞刺可能对ALS患者有一定疗效。但临床上应用大剂量环磷酰胺治疗ALS却未能改变病程,免疫治疗仍需要更进一步的研究。
干细胞移植治疗:
干细胞是指可以在体外大最扩增并具有多项分化潜能的细胞。干细胞移植是现在研究的热点。许多动物实验研究表明脐血干细胞可以延长ALS小鼠的生存期。神经干细胞是近年来的重大发现,其比胚胎干细胞更加成熟,能集中迁徙至损伤部位。目前已经开始临床干细胞治疗ALS,很多患者症状改善,肌肉萎缩好转,肌肉力量增加,生存期延长。干细胞治疗,是目前治疗ALS的有效方法,虽不能治愈ALS,却可以显著提高ALS患者的生存质量。
支持治疗:
适当功能锻炼是有益的,切勿剧烈采用物理及运动疗法。均衡饮食,吞咽困难者宜摄取高蛋白、高热量的糊状饮食,严重者可鼻饲。呼吸困难者宜使用呼吸机,可及时清除呼吸道分泌物以防止窒息。双水平正压通气(BIPAP)能主动辅助患者的吸气相,早期使用能显著延长生存期。经皮内窥镜胃造瘘(PEG)的胃肠营养可以显著延长生存期。
综合治疗:
在ALS病程的不同阶段,患者所面临的问题有所不同,如抑郁焦虑、失眠、流涎、构音障碍、交流困难、肢体痉挛、疼痛等,应根据患者具体情况,给予针对性的指导和治疗,选择适当的药物和辅助设施,提高生活质量,加强护理,预防各种并发症。
接诊ALS病人的个人体会
在我们接诊的125例病例,男女比例为1.6:1,多数为中年后起病,最小发病年龄32岁,最大发病年龄59岁,平均发病年龄为54岁,均为家族中首例发病患者。
起病的首发症状多为单侧或者双侧肢体无力,上肢起病居多,1/3患者以吞咽困难起病,所有患者均有不同程度的肌肉萎缩,来我们院治疗前的病程在6个月至3年,均伴随有不同程度的抑郁。
在众多的病例中,有3例患者给我们的印象尤为深刻。
第1例患者是为55岁的女性,来我们院前10个月无明显诱因出现左侧拇指痉挛疼痛,就诊当地医院行颈椎CT检查,结果回报示:颈椎3、4、5、6椎间盘膨出,医生考虑“颈椎病”,建议手术治疗,患者未接受手术治疗的方案,转另一家医院就诊,行神经系统检查,初步诊断为“肌萎缩侧所硬化”,坚持中医中药、针灸及抗抑郁治疗,病情逐渐加重,左上肢明显肌肉萎缩,吃饭、梳头、洗澡困难,咳嗽、打喷嚏无力,体重减轻6公斤,精神差,饮食差,睡眠差。在我们院治疗5周后患者萎缩的肌肉不同程度的恢复,左上肢力量较前提升20%,出院后1个月患者因呼吸衰竭死亡。
第2例患者是位59岁男性,2006年无明显诱因出现行走速度缓慢,无震颤、平衡障碍、肌肉萎缩等异常,未行诊治;病情进行性加重,在1年半内病情加重较快,开始出现行走困难,吃饭、穿衣、刷牙、洗脸困难,需要别人帮助才能完成,在当地医院就诊未明确诊断。至2009年患者四肢无力进行性加重,伴有明显肌肉萎缩,并伴有吞咽困难、呼吸困难、书写困难及饮水呛咳等症状,活动耐力差,行肌电图后诊断为运动神经元病,服用利鲁唑、巴氯酚、维生素E等治疗,效果不佳,2009年7月开始在我们院治疗,治疗后患者依靠助行器可以行走,吞咽困难、呼吸困难未再加重,老先生很乐观,坚持每年治疗一次,病情进展缓慢。
第3例患者也是一位59岁的男性,病史1年,右手无力及轻微吞咽困难起病,至当地医院就诊,历时半年,确诊为“肌萎缩侧索硬化症”,未治疗,患者右手无力逐渐加重,逐渐累及右上肢、右下肢及左上肢,吞咽困难逐渐加重,并出现言语缓慢,时有吐字不清,在我们院治疗后患者左侧肢体力量恢复正常,右侧肢体力量接近正常,吞咽困难不明显,说话吐字明显清晰。
通过这3个病例故事,我们想说的是,对于ALS这个疾病,没有经历过或者见过的人真的没办法理解它的可怕,它就像慢性毒药一样一点点吞噬着患者的生命。每天我们面对这样的病人,感受着他们的痛苦,尽最大努力的去缓解他们的病痛,通过药物及心理治疗改善患者的抑郁状态,我们鼓励每一个患者及家属不抛弃不放弃,保持积极乐观的态度,这对于病情的恢复有莫大的帮助;作为一个神经内科医生,我们要求自己给予这类患者更多的人文关怀,因为他们经历的病痛是一般人无法想象的。通过这3个病例我们还可以看到,早诊断早治疗是非常有必要的,治疗的效果与病程、病情进展速度和患者的情绪状态等有一定关系的。作为神经内科的疑难杂症,ALS值得全世界关注。(编辑:粉条er)
参考文献:
谢圣瑞.肌萎缩侧索硬化症治疗的研究进展[J].中国康复理论与实践,2010,16(6):535-537
神经病学(第6版)
中华医学会神经病学分会.肌萎缩侧索硬化的诊断标准(草案)[J].中华神经科杂志,2001,34(3):190
Lacomblez L,Bensimon G,Leigh P N,et aI.Dose_ranging study of riluzole in amyotrophic lateraI sclerosis.Amyotrophic Lateral sclerosis/Riluz01e study Group ll[J].Lancet,1996,347(9013):1425一1431.
中华医学会神经病学分会肌电图与临床神经电生理学组,中华医学会神经病学分会神经肌肉病学组.中国肌萎缩侧索硬化诊断和治疗指南[J].中华神经科杂志,2012,45(7):531-533
Estevez AG, Crow JP,Sampson JB.et a1.1nduction of nitric oxide—dependenl apoptosis in motor neurons by zinc—deficient superoxide dismutase[J].science, 1999。286(5449):2498—2500.
Haman D.The free radical theory of aging[J]. AntioxidRedox Signal·2003。5(5):557—561.
Graf M,Ecker D, Horowski R, et a1.High dose vitamin E therapy in amyotrophic lateral sclerosis as add—on therapy to riluzole:results of a placebo—contro¨ed double—b“nd study[J].J Neural Transm。2005,112(5):649—660.
Ascherio A,Weisskopf MG,OReilly EIJ,et a1.Vitamin E intake and risk of amyotrophic lateral sclerosis[J]. Ann Neuml,2005,57(1):104一110.
Aoyama N,Katayama Y,Kawamata T,et a1. Effects of antioxidant,OPC-14117,on secondary cellular damage and behavioral deficits following cortical contusion in the rat[J].Brain Res,2002,934(2):117—124.
Abe K, Morita S, Kikuchi T,et a1.Protective effect of a novel free radical scavenger,()PC-14117,on wobbler mouse motor neuron disease[J].J Neurosci Res。1997,48(1):63—70.
Szczudlik A,Tomik B, Slowik A,et aI.Assessment of the efficacy of treatment with pimozide in patients with amyotrophic lateral sclerosis. Introductory note5[J]. Neurol Neurochir Pol,1998,32(4):821—829.
Mitsumoto H,lkeda K,Holmlund T.et a1.The effects of ciliary neurotrophic factor on motor dysfunction in wobbler mouse motor neuron disease[J].Ann Neurol,1994。36(2);142—148.
Al-chalabi A,scheffler MD,smith BN,et a1.Ciliary neurotrophic factor genotype does not influence clinical phenotype in amyotrophic lateral sclerosis[J].Ann Neur01.2003,54(1):130一134.
Ilzecka J. Increased serum CNTF level in patients with amyotrophic lateral sclerosis[J].Eur cytokine Netw,2003,14(3):192—194.
Alisky JM, Davidson BL. Gene therapy for amyotrophic lateral sclerosis and other motor neuron diseases. Hum Gene Ther,2000 , 11(17): 2315 22329.
Hayata N,Fujio Y,Yamamoto Y,et a1.connective tissue growth factor induces cardiac hypertrophy through Akt sig—naling[J].Biochem Biophys Res commun,2008。370(2):274—278.
DAmelio P,Cristofaro MA, Tamone C,et a1.Role of iron metabolism and oxidative damage in postmenopausal bone loss[J].Bone,2008,43(6):1010—lol5.
Ozgocmen s,Kaya H,Fadillioglu E,et a1.Role of antioxidant systems,lipid peroxidation,and nitric oxide in postmenopausal osteoporosis[J].M01 Cell Biochem,2007,295(1—2):45—52.
Maziere c,I,ouvet L,Gomila c,et a1.Oxidized low density lipoprotein decreases Rankl—induced differentiation of osteoclasts by inhibition of Rankl signaling[J].J Cell Physiol,2009,221(3):572—578.
Banfi G,Iorio EL,Corsi MM.oxidative stress,free radicals and bone remodeling[J].clin Chem Lab Med,2008,46(11):1550一1555.
崔芳.肌萎缩侧索硬化症的研究进展[J].实用临床医药杂志,2010,14(11):104-107
文章题图:macleans.ca