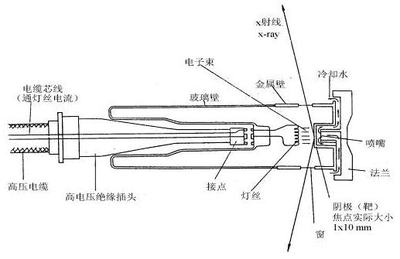
利用单晶体对 X射线的衍射效应来测定晶体结构的实验方法。依照强度记录方式的不同,可分为照相法和衍射仪法两类。
x射线衍射_单晶X射线衍射 -单晶X射线衍射
x射线衍射_单晶X射线衍射 -正文
利用单晶体对 X射线的衍射效应来测定晶体结构的实验方法。依照强度记录方式的不同,可分为照相法和衍射仪法两类(见彩图)。单晶X射线衍射照相法使射线作用在胶片上,然后测量底片上衍射点的黑度来获得衍射线的强度数据,根据实验装置和条件的差别,又分为多种方法。
劳厄照相法用连续波长的 X射线照射到静止不动的单晶体上,通常采用平板底片,所摄得的衍射图称为劳厄图。劳厄图常用来测定晶体的对称性和用于晶体的定向等。
转动照相法单波长的 X射线垂直照射到不停地转动着的单晶体上,底片卷成圆筒状并使其轴线与晶体的转轴重合。若转轴与晶体的某一晶轴(假定是c轴)平行,则衍射线分布在如图1a
所示的一系列圆锥面上,衍射图展平后如图1b所示,图中衍射点排列成一系列平行的层线。利用转动图中的层线间距可算出晶格参数(见点阵),若晶体的c轴与转动轴一致,则c轴长可由下式求得:式中λ为X射线的波长;r为圆筒形底片的半径;yn为第n层层线和零层线的距离。对于结构简单的晶体,转动法可用于测定其晶体结构;但对于较复杂的结构,转动法就有困难,这是因为同一层线的衍射点分布密集,不利于衍射点的指标化和强度测量。
回摆照相法实验条件和装置与转动法基本一样,差别在于照相过程中,晶体只在选定的角度范围内来回摆动。这样可以避免同一层线上衍射点的重叠。但要摄取多套回摆图,才能收集完整的衍射数据。回摆照相法若配上计算机,自动测量衍射点强度和指标化,则有相当的优越性。目前广泛用于蛋白质的结构分析,与四圆衍射仪比较,可节省衍射实验的时间。
韦森堡照相法同一层线的衍射点是由不同晶面在晶体转动的不同时刻反射得到的,若在晶体转动时,让带动晶体摆动的马达通过涡轮涡杆同时使底片圆筒左右来回摆动,就可将原在同一层线的衍射点分开,这类方法称为运动底片法。韦森堡照相法是运动底片法的一种,其装置如图2所示。晶体的转轴和感光胶片圆筒均水平安放,在晶体与底片之间有一个层线屏,以便将其他层的衍射线遮住,只让某一层线的衍射线射到底片上,这类衍射图如图3所示。
单晶X射线衍射
单晶X射线衍射韦森堡图既可用来确定晶体的微观对称性和晶格参数,又可较方便地进行衍射点的指标化和测量强度,因而在四圆衍射仪被广泛使用之前,它是测定晶体结构的重要方法之一。
单晶衍射仪法此法用射线计数仪直接记录射线的强度。单晶衍射仪有线性衍射仪、四圆衍射仪和韦森堡衍射仪等,其中以四圆衍射仪(图4),(见彩图)最为通用。所谓四圆是指晶体和计数器藉以调节方位的四个圆,分别称为φ圆、χ圆、w圆和2θ圆。φ圆是安装晶体的测角头转动的圆;χ圆是支撑测角头的垂直圆,测角头可在此圆上运动;w圆是使χ圆绕垂直轴转动的圆,2θ圆与w圆共轴,计数器绕着这个轴转动。这四个圆中,w圆、φ圆和χ圆用于调节晶体的取向,使某一指定的晶面满足衍射条件,同时调节2θ圆,使衍射线进入计数器中。通常,四圆衍射仪配用电子计算机自动控制和记录,可以精确测定晶格参数,并将衍射点的强度数据依次自动收集,简化了实验过程,而且大大提高了数据的精确度。因此,它已成为当前晶体结构分析中强有力的工具。
x射线衍射_单晶X射线衍射 -晶体衍射的基本公式
由于晶体中原子是周期排列的,其周期性可用点阵表示。而一个三维点阵可简单地用一个由八个相邻点构成的平行六面体(称晶胞)在三维方向重复得到。一个晶胞形状由它的三个边(a,b,c)及它们间的夹角(γ,α,β)所规定,这六个参数称点阵参数或晶胞参数,见图1。这样一个三维点阵也可以看成是许多相同的平面点阵平行等距排列而成的,这样一族平面点阵称为一个平面点阵族,常用符号HKL(HKL为整数)来表示。一个三维空间点阵划分为平面点阵族的方式是很多的,其平面点阵的构造和面间距d可以是不同的。晶体结构的周期性就可以由这一组dHKL来表示。
一个小晶体衍射X射线,其衍射方向是与晶体的周期性(d)有关的。一个衍射总可找到一个晶面族HKL,使它与入射线在此面族上符合反射关系,就以此面族的符号HKL作为此衍射之指数。其间关系用布拉格方程(式1)来表示。
2dHKLsinθHKL=nλ(1)
式中,θHKL为入射线或反射线与晶面族之间的夹角,λ为入射X射线波长,n为反射级数。
衍射线的强度是与被重复排列的原子团的结构,也即和原子在晶胞中的分布装况(坐标)有关,其间的关系由方程式(2)表示
式中,E称为累积能量,I0为入射线强度,e,m为电子的电荷与质量,c为光速,λ为X射线波长,Vu为晶胞体积,称洛仑兹偏振(LP)因子,|F|为结构振幅,e-2MT为温度因子,A为吸收因子,V为小单晶体的体积,ω为样品的转速,其中结构因子=|FHKL|eiαHKL(3)
式中,fj,xj,yj,zj分别为第j个原子的原子散射因子及它在晶胞中的分数坐标(以晶胞边长为1)。n为晶胞中的原子数。αHKL为HKL衍射的相角。从此式可知衍射线强度是与各原子在晶胞中的位置(即结构)有关的,故反过来可从衍射线强度的分析解出晶胞中各原子的位置,即晶体结构。其方法是(4)
通过晶胞中的电子密度ρ(x,y,z)的计算。
故若知各衍射的FHKL,就可按(4)式计算晶胞的三维电子密度图。原子所在处电子密度应该很高,故依此可定出原子在晶胞中位置,得出晶体结构。但是从衍射强度获得的是结构振幅|F|,|F|与F之间的关系见式(3)。如何求得各HKL衍射的相角αHKL就成为X射线单晶衍射解晶体结构的关键。
x射线衍射_单晶X射线衍射 -可能的发展方向
数据的积累
从前述的应用已经看出,晶体结构的测定及结构与性能关系的研究,是今后走上人类按需设计新材料的基础。今日虽已测了许多晶体的结构,但还有许多未能测定,而且还不断有新化合物,新晶体出现,因此不断的测定他们的结构,加以总结分析是十分必要的。当今已有多个晶体结构数据库,如:(1)剑桥结构数据库(CSD)。包括各种有机,有机金属化合物及配合物的晶体学数据。(2)无机晶体结构数据库(ICSD)。(3)金属结晶学数据库(CRXSTMET)。(4)蛋白质数据库(PDB)。(5)晶体学数据(CD)。(6)粉末衍射卡片(PDF)。(7)有序无序结构数据库(OD)等。
各种生物大分子结构的测定
2001年2月12日,人类基因组框架图发表。接下来的任务是要把各基因的结构和功能搞清楚,有大量的基因结构需要测定。世界上已经成立了结构基因组的国际合作组织,分配人类基因结构的测定任务。除了人类基因以外,还有水稻基因组,各种病毒等范围更广的生物大分子结构需测定。生物大分子的数量将会远远超过各种无机物,有机物分子的总量。生物大分子结构测定将是今后晶体结构测定的主要任务。
结构与性能关系的研究与应用
按照已知的结构和性能的关系设计制造需要的新材料是进行大量结构测定的目的。如何总结大量已测结构的规律并与其性能、功能相联系是今后的任务之一。特别是生物结构与功能的关系。进一步如何利用这种关系设计制造人类需要的材料,药物等,更是永不完结的任务。
解生物大分子结构方法的发展
目前虽已有各种方法用来解决相角的问题,但要置换许多同晶化合物还是颇费时和颇昂贵的,如果能如小分子那样用直接法来解决相角问题,将会方便许多。我国科学家范海福院士是研究直接法的世界权威人物,正在进行这方面的研究。
单晶体结构分析实验方法的发展
目前的实验室单晶体结构分析方法对于测定小分子的单晶体结构已经是相当完美了,但对于巨大的生物大分子就显得软弱无力,主要是光源强度不够,光的平行性不良,波长又不好调。目前主要要依靠同步辐射作为X射线源。我国二个同步辐射光源之一的位于合肥的国家同步辐射实验室(NSRL)今年上半年已胜利完成用于生物大分子结构测定的光束线与实验站的建设,并已收集到尖吻蝮蛇蛇毒磷脂酸A2,神经毒等多种生物大分子的衍射数据,并解出了结构。另一个北京同步辐射装置(BSRF)的生物平台使用的是聚焦光束。也已于2002年12月4日投入试运转,在样品冷冻条件下成功地收集了18套结构数据,情况良好。数据分析正在进行中。
同步辐射

是一种大科学装置,设备大投资高,一般都需要政府投资,不是一般实验室所能具备的,需要申请立项才能使用。因此,如果能发展出高强度的实验室光源和极高灵敏度的探测器,使在一般实验室中也能测定生物大分子结构,则绝对是有益的。
有许多生物反应的速度是相当快的,如血红蛋白与一氧化碳的结合,速度在纳秒级(10-9sec),要对这种反应进行动力学研究,既要有高强度脉冲光源,又要有快速切换的探测器以连续跟踪反应。现在已有了强脉冲光源,但探测器的切换速度却慢太多,需要作长时间的更大的努力。
x射线衍射_单晶X射线衍射 -编辑本段衍射数据的处理
一般步骤
1.选择大小适度,晶质良好的单晶体作试样,收集衍射数据。
2.指标化衍射图,求出晶胞常数,依据全部衍射线的衍射指标,总结出消光规律,推断晶体所属的空间群。
3.将测得的衍射强度作吸收校正,LP校正等各种处理以得出结构振幅|F|。
4.相角和初结构的推测。常用推测相角的方法有派特逊函数法及直接法。
派特逊函数的定义
从派特逊图上可以比较容易地得到晶体中所含重原子的位置坐标,可依此计算各衍射的相角αHKL,将此αHKL去与实验测得的结构振幅|FHKL|结合生成FHKL,可据此计算电子密度图,可以定出更多原子的原子位置及修正已有的原子位置。再利用这些数据重新计算αHKL、FHKL及ρ(x,y,z),如此反复叠代多次推出完整的晶体结构。
直接法是利用结构振幅间的某些统计关系求出衍射相角的方法。在求出某些衍射的相角αHKL以后,把他们去与实验测得的|FHKL|配合生成FHKL,进而计算ρ(x,y,z),从中获得部分原子的位置,从此修正和扩充已有的相角,如派特逊那样反复叠代以得出完整的结构。
结构的精修
由派特逊函数或直接法推出的结构是较粗糙和可能不完整的,故需要对此初始结构进行完善和精修。常用的完善结构的方法称为差值电子密度图,常用的精修结构参数的方法是最小二乘方法,经过多次反复,最后可得精确的结构。同时需计算各原子的各向同性或各向异性温度因子及位置占有率等因子。最终所得结果的优劣常用吻合因子R来衡量式中,w为权重因子,下标o,c表示实测值,计算值。
结构的表达
在获得精确的原子位置以后,要把结构完美的表达出来,这包括键长键角的计算,绘出分子结构图和晶胞图,并从其结构特点探讨某些可能的性能。
生物大分子结构的测定
生物大分子结构的测定,从原理上讲与小分子(无机物,有机物,配合物等)无异,但由于分子大也带来一定的特殊性,需要有一些不同于小分子的方法。
大分子的特点是分子量大,原子多,但原子序数小,多数是C,H,O,N等轻元素,故散射能力低,不易收集到高角的衍射点,这会使电子密度图的分辨率降低。还因他晶胞大,所含原子数目多,需确定的结构参数就多,这与分辨率低有矛盾。另外,大分子晶体在X射线的照射下易于损坏,如何缩短实验时间也成为一个重要问题。
大分子由于分子量大,要求使用较大体积的单晶体,如对于分子量为5000的蛋白质分子,就要有大于0.3mm3的单晶体。但因其分子大,结晶就困难,而且很易结晶成孪晶,这不合用。得到合用的晶体是整个大分子结构测定中关键的一步,常说有了良好的晶体,结构测定一半的问题已解决了。
一般用回摆法收集衍射数据,用IP或CCD作探测器,可以在几个小时内完成数据收集工作。
关于相角问题,在解小分子结构中目前最常使用的直接法还不能用于大分子,而派特逊法,由于大分子缺少重原子也无法使用。目前是用基于派特逊法的几个方法来解决相角问题的。
分子取代法
有时,几个不同的生物大分子是由相同的结构单位以不同方式连接而成的;同一种生物大分子如在不同的条件中结晶,有可能得到不同的结晶,是为多结晶现象;还有些生物分子,他们是由共同的分子祖先进化而得,因此其中有相当部分是相同的。对于这些类的生物分子,他们的派特逊图常有很大的相似性。因此,在求解某生物大分子结构时,如能找到与他有类似的结构,且结构已测定的生物大分子结构时,则可按最大重叠原理找出待测物和已知物的派特逊图之间的关系,从而得出未知物衍射的位相,进而解出结构。若已知物和待测物是同晶化合物,则可利用差值电子密度图来解出结构,更为简单。
若在已知结构的大分子结构数据库中找不到与待测物有类似结构的分子结构时,就不能使用此法,要使用以后的方法。
重原子同晶置换法
这种方法是设法把对X射线散射能力大的重金属原子,如Hg,Pb,Se等引入生物分子中,作为标识原子。这种置换入重原子的大分子应与无重原子时的原晶体有相同的晶胞参数和空间群,且绝大多数原子的位置相同,故称同晶置换。从这些含重原子晶体的衍射数据,利用基于派特逊法的方法可解出重原子的位置,据此算出其结构因子和相角,进而利用相角关系计算出没有重原子的原晶体的相角,解出结构。
经常使用不只一种重原子进行置换,以得几种同晶置换衍生物,称多对同晶置换法。同晶置换,衍生物越多,可正确定出的相角也越多。
反常散射法
晶体衍射中有一条弗里德耳定律,就是说不论晶体中是否存在对称中心,在晶体衍射中总存在着对称中心,也即有FHKL=FHKL。但是当使用的X射线波长与待测样品中某一元素的吸收边靠近时,就不遵从上述定律,也即FHKL≠FHKL。这是由电子的反常散射造成的,利用这一现象可以解决待测物的相角问题。一般,这一方法常与重原子同晶置换法结合使用。
在收得同晶置换物的衍射数据后,改变入射线波长至靠近重原子的吸收边处,再次收集数据,这套数据是存在反常散射的,可利用这两套数据来求位相。有如多同晶置换法,如采用几个不同波长的X射线,对所含不同元素收集几套反常散射数据,则可得更正确、更完整的相位信息,是为多波长反常衍射法(MAD),是目前解分子生物大分子的重要方法。实验室X射线光源不易使用这一方法,因要改变X射线波长是很困难的,而对于同步X射线光源,改变波长是相当方便的,因此常被使用。