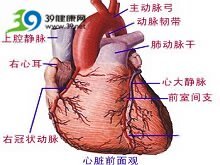
gardner综合征_Peutz-Jeghers综合征 -特征

本征有3大特征:①黏膜、皮肤特定部位色素斑;②胃肠道多发性息肉;③遗传性。以往认为本病罕见,近年来临床报道病例较多。本病可发生于任何年龄,多见于儿童和青少年,男女发病率大致相同。
1896年Hutchinson首先报道了1对孪生女孩的上唇有黑色素斑点,1919年其同事Weber报道了两姊妹中的1个在20岁时死于肠套叠。1921年Peutz报道了一家三代人中曾有7人患小肠息肉病,口唇和颊黏膜有黑色素斑点。描述了本病的家族遗传性。1949年Jeghers会同Mckusick和Katz等收集了世界文献22例,并报告了自己的10例,强调了本病的家族遗传性及皮肤、黏膜色素斑的特点,引起了广泛注意。1954年Bruwer等首次使用Peutz-Jeghers综合征这一名称。Vilchis等报道,此综合征全世界每年平均有10例。但1977年Mcallister等收集欧美文献共320例,1987年后藤明彦收集日本文献共355例,孟荣贵等收集1985-1989年国内文献44篇共173例,说明pjs在我国并不少见。
gardner综合征_Peutz-Jeghers综合征 -一.病因
PJS属于家族遗传性疾病,其遗传方式为常染色体显性遗传,由单一多效基因所传递。患者的染色体分纯合子和杂合子2种,由于基因的突变,二者都能发病。纯合子的出现率很低,而往往易致死胎或夭亡。在临床上所见的患者中以杂合子居多。在双亲中的一方正常,另一方为杂合子,其子女中约有1/2可能发病。患者的健康子女如果不是近亲婚配,不会有致病基因传给后代而发病。郭敏等报道了3个家族8例患者中,一家为父女遗传;一家为母子遗传;另一例共生4子,第3、4子则患本病,其长子仅唇部、口腔黏膜有黑色素斑,并无胃肠道多发性息肉,可称为不完全的显性遗传。PJS患者约有50%无明显家族史,可能是由于新的基因突变所造成的,但其后代仍有发病的可能。目前还不能通过遗传标志预测本病患者子女中谁可能发病,这有待进一步研究。gardner综合征_Peutz-Jeghers综合征 -二.病理
PJS患者的主要病理改变为黏膜、皮肤色素斑和胃肠道息肉。黏膜、皮肤色素斑为真皮基底内黑色素细胞数量增加,黑色素沉着所形成。本征的息肉为错构瘤性,非肿瘤性息肉。息肉的表面是正常肠或胃上皮细胞所构成的腺管。