地中海贫血(Thalassemia),又称海洋性贫血,简称地贫。是一组遗传性小细胞性溶血性贫血。其共同特点是由于珠蛋白基因的缺陷使血红蛋白中的珠蛋白肽链有一种或几种合成减少或不能合成。导致血红蛋白的组成成分改变,本组疾病的临床症状轻重不一,大多表现为慢性进行性溶血性贫血。
地中海贫血基因_地中海贫血症 -简介
地中海贫血症
地中海贫血,是一种先天的血液疾病,和父母的遗传有关。1925年意大利首次报道了此病,目前该病已在全世界范围散布。患者的红血球较脆弱且容易死亡,其带氧能力亦不足,超过某种程度无法正常生活,不能有效地制造红血球,因而长期有溶血性贫血的现象。在结婚以前健康检查可以筛选出来,是一种隐性基因遗传,患者红血球的体积较正常细胞小,且有时因血红素含量低较苍白或呈靶型(targetcells)。由于此病流行于地中海、中东及东南亚一带地区,故被称为“地中海贫血”。
地中海贫血基因_地中海贫血症 -历史
1925年,意大利医生Cooley首次报道5例具有严重贫血、脾脏肿大、异常的骨骼病变以及外周血出现大量幼稚红细胞的小儿病例。
1932年,Whipple和Brasford借用希腊语的单词(Thalassemia)即是“海”的意思,Hemia即是“血”的意思)将地中海贫血定义为海泮性贫血。
1940年,美国血液学家Wintrobe报道Cooley贫血患者的家族中存在轻型地中海贫血患者,指出地中海贫血系常染色体不完全性显性遗传,有纯合子与杂合子之分。
地中海贫血基因_地中海贫血症 -分布
地中海沿岸国家:如意大利、希腊、塞浦路斯。
非洲:主要在北非国家,阿尔及利亚及利比亚北部、埃及、苏丹和索马里北部。
亚洲:中东国家(沙特阿拉伯、伊朗、伊拉克)、中国、东南亚各国(印度、巴基斯坦、印度尼西亚、马来西亚等)。
中国:长江以南省份,尤其以广东、广西、海南、四川、云南、台湾等省份为多发。
地中海贫血基因_地中海贫血症 -发病率
不同种族的人群中地中海贫血基因频率及发病率:
在意大利卡拉里奇地区、西西里岛人群中,β-地中海贫血杂合子约为10%。意大利撒丁岛β地贫基因携带率约为1/8,夫妇双方均为携带者机率1/64,重型β-地贫发生率为1/258。希腊7.0%,塞浦路斯15.0%。
东南亚:印度3.7%,巴基斯坦4.0%,泰国4.8%。
中国:云南:4.8%、四川:2.37%、贵州:2.21%,福建:1.83%、广西:1.52%、广东:1.08%。
地中海贫血基因_地中海贫血症 -病因
本病是由于珠蛋白基因的缺失或点突变所致。根据其变异的肽链可分为α地贫和β地贫。有可能是因为近亲通婚所产生的遗传疾病。
地中海贫血基因_地中海贫血症 -类型
地中海贫血症
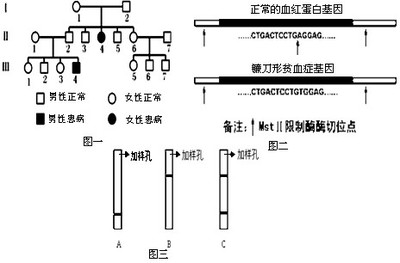
根据血红蛋白中不同位置的损害可分成两类:α地中海贫血与β地中海贫血。α地中海贫血是血红蛋白中的α血红蛋白链有缺损,β地中海贫血则是血红蛋白中的β血红蛋白链有缺损。镰刀型红血球疾病,又名镰刀型贫血(SickleCellAnemia)和地中海贫血不同的是它只发生β血红蛋白的缺陷。
α地中海贫血
α地中海贫血的患者会在HBA1、HBA2这两个基因发生异常。在成人造成β蛋白链过度制造,在新生儿则是γ蛋白链过多。过多的β蛋白链形成四聚物(tetramers)使红血球的携氧能力降低。
β地中海贫血
β地中海贫血的患者则是在第11号染色体上的HBB基因发生突变。不正常的细胞会制造过量的α蛋白链,然后结合在红血球的细胞膜上造成细胞膜损坏;若其浓度过高,则有形成有毒聚集体(aggregates)的可能。

地中海贫血基因_地中海贫血症 -症状
地中海贫血症
地中海贫血症有隐性、轻型和重型之分。重型患者需要终生定期的输血和接受药物治疗;而两个隐性或轻度患者结婚,他们的下一代则有1/4机会患有重度地中海贫血症。相反地,两者中只有一位是存有地中海贫血症基因的话,不论程度如何,则下一代没有此问题或只带有隐性或轻度。
由于地中海型贫血的患者缺少正常的血红素,红血球携氧功能差,体内主要造血器官骨髓与次要造血器官肝脏、脾脏均会进行旺盛的造血作用,但造出的红血球也多半品质不佳,容易被破坏,成为恶性循环。骨髓增生会侵犯周围的皮质骨,使骨骼较脆弱。旺盛的造血作用会消耗极多的养分与能量,使身体其他部位的养分供需失调。不断的输血可以改善贫血的症状,也可避免过度的造血作用,但血红素中的铁质会过度存在身体中,并堆积至各重要器官造成器官病变。
而生活中,地中海贫血患者因血红素带氧量不足而影响患者在体力上的差异,患者不适宜进行太激烈的运动,还需要打除铁针去除体内多余的铁质。另外亦因为血红素不足的关系,患者比较容易有头晕、头痛甚至腰痛的症状。
地中海贫血基因_地中海贫血症 -并发症
过量铁质积聚
长期输血会造成铁质沉积而过量铁质的积聚会对多个器官造成破坏。主要受影响的包括心脏、肝脏、胰脏和各个内分泌器官。病者会出现心脏衰竭、肝硬化、肝功能衰退、糖尿以及因为多种内分泌失调而变得身材矮小和发育不全等等。
输血引起不良反应
常见输血时引起的不良反应包括发热、发冷和出红疹等。较严重的反应如急性溶血、气管收缩和血压下降等虽然甚少出现,但绝不能忽视。在长期贫血和溶血的刺激下,不少重型和中型贫血病者都会出现脾脏发大的问题。过大脾脏会使贫血加剧和令病者需要接受更大量的输血而导致更严重的铁质积聚。及时把发大的脾脏切除往往能令情况改善。长期溶血令地中海贫血病人比一般人更容易患胆石。患有胆石的病人可能经常出现右上腹痛、皮肤、眼白变黄和茶色小便等的病征。
除铁药的副作用
除铁药会影响视力、听觉和骨骼生长。
地中海贫血基因_地中海贫血症 -诊断
实验室检查:外周血象呈小细胞低色素性贫血,红细胞大小不等,中央浅染区扩大,出现异形、靶形、碎片红细胞和有核红细胞、点彩红细胞、嗜多染性红细胞、豪?周氏小体等;网织红细胞正常或增高。骨髓象呈红细胞系统增生明显活跃,以中、晚幼红细胞占多数,成熟红细胞改变与外周血相同。红细胞渗透脆性明显减低。HbF含量明显增高,大多>0.40,这是诊断重型β地贫的重要依据。颅骨X线片可见颅骨内外板变薄、板障增宽,在骨皮质间出现垂直短发样骨刺。
地中海贫血基因_地中海贫血症 -治疗
现时的移植疗法是从兄弟姊妹抽取骨髓、脐带血或血液中的干细胞移植到病人身上。若移植成功,病人的骨髓便回复正常的造血功能,贫血得以痊愈。移植疗法有一定危险性,病人在治疗过程中亦可能要忍受相当的痛苦。
地中海贫血基因_地中海贫血症 -预防
1、开展人群普查和遗传咨询、作好婚前指导以避免地贫基因携带者之间联姻,对预防本病有重要意义。采用基因分析法进行产前诊断,可在妊娠早期对重型β和α地贫胎儿作出诊断并及时中止妊娠,以避免胎儿水肿综合征的发生和重型β地贫患者出生,是目前预防本病行之有效的方法。
2、地中海型贫血带因者外表、成长与正常人无异。
3、维他命之补充同平常人,于医师认为必要时投予。因外表不易查觉此病,常被给予铁剂补血,应注意。
4、婚前检查:结婚对象应检验是否为地中海型贫血带因者,若是则特别注意产前检查。
5、产前检查:若夫妻均为带因者,每胎怀孕第12周以后即应抽取胎儿检体检,若确定为地中海型贫血重型胎儿即可予人工流产,以免将来照顾上的负担。
地中海贫血基因_地中海贫血症 -相关报道
中国发现世界首例缺失型α地中海贫血突变基因
中国医务人员发现世界首例缺失型α地中海贫血突变基因,并于日前(2013年11月)在美国DNA数据库成功注册。这一新突变基因的发现,不仅丰富了世界地中海贫血突变基因数据库,同时为在临床上避免地贫患儿的降生及科学研究提供了新的参考信息。
据介绍,该例缺失型α地中海贫血突变基因由李友琼等医务人员于2011年首次发现,携带者是当时到该院做产前检查的一名孕妇。经过血红蛋白电泳、地贫基因筛查分析和相关医学科研院校的研究,并借助基因公司深度测序平台的检测手段,最终于2012年底完成基因的鉴定工作。
罕见病百科行业百科
概述病例资讯科学研究治疗相关孤儿药信息会议培训医师、专家行业动态药物药品中国地区外国药物政策法规立法国家救助疾病词条罕见病词条分类热搜罕见病词条常见罕见疾病未分类或不明原因中外对比交流社区公益组织交流人群得了罕见病,究竟该去哪里治?疑难病、罕见病不能轻易断定,医生需要反复思考、检查,留意观察病情,对于实在不能解决的疾病才能称为疑难病或罕见病。所以,他说,医生在碰到少见或者有点棘手的疾病时,不要轻易下判断,要从常见病或者多发病着手处理。
瓷娃娃(成骨不全症)疾病简介患有成骨不全症(OI)的儿童以及他们的家庭所面临的问题是复杂的,涉及到解剖、医疗、对残疾的适应和社会等多个层面。其中某些问题是难以克服的,可能无法彻底解决……
哪天是国际罕见病日?由欧洲罕见病组织(EURORDIS)于2008年发起,确定2月29日为国际罕见病日,以这个四年一次的日子意寓罕见病之“罕见”。
什么是罕见病?罕见病,是指盛行率低、少见的疾病,在美国罕见疾病组织所公布的罕见疾病高达一千种之多。
为什么Mayo Clinic这么牛?梅奥医院(英语:Mayo Clinic),是世界著名的医疗机构,位于美国明尼苏达州罗彻斯特(Rochester)。它还有医院设在佛罗里达州的杰克逊维尔(Jacksonville)及亚利桑那州的斯科茨代尔(Scottsdale)。在明尼苏达州、艾奥瓦州、威斯康辛州还有一些小的诊所和医院。
查看“罕见病百科”更多内容>>