嗜酸性粒细胞增生性淋巴肉芽肿以淋巴细胞和嗜酸性细胞增生、浸润为主要病理特点,以淋巴结肿大,软组织肉芽肿形成为主要临床表现,病因不明。
嗜酸性淋巴肉芽肿即Kimura's病(KD)是一种少见的、原因不明的、多累及头颈部浅表淋巴结及软组织的慢性肉芽肿病变。1909年日本学者片山最早发现此病,但因年代久远而资料不全。我国学者金显宅于1937年也曾有报道“嗜伊红血球增多性淋巴母细胞瘤”,后发现为非肿瘤性病变,又于1957年更名为“嗜酸性淋巴肉芽肿”。1948年日本另一学者木村哲二(Kimura)对此病作了详细报道,引起学术界重视,之后此病又被命名为KD。
此病命名曾较混乱,有“类似慢性肉芽肿伴嗜酸性细胞增多症”、“软组织嗜酸性肉芽肿”、“嗜酸性淋巴滤泡病”、“皮肤嗜酸性淋巴滤泡增多症”、“淋巴结和软组织的嗜酸性肉芽肿”、“嗜酸性细胞增多性淋巴肉芽肿”等病名。目前国内文献多采用“嗜酸性淋巴肉芽肿”,而西方及日本的文献主要用“KD”。
该病少见,至今报道仅300例,主要见于口腔、皮肤及放疗等专业的文献,耳鼻咽喉科医生报道此病的甚少。因本病的主要症状之一为头颈部肿块,与耳鼻咽喉科关系密切,应为广大耳鼻咽喉科医生所认识。
1病因
目前尚不明确。现已证实与TB、梅毒、化脓菌、霉菌、病毒无关。因病变组织内有大量嗜酸性粒细胞浸润,外周血嗜酸性粒细胞明显升高,血清IgE升高,且可合并肾病综合征及支气管哮喘并发症,因此多数学者认为该病是一种免疫介导的炎性反应性疾病。杨洋等〔1〕在KD患者的组织切片中还发现有肥大细胞增生及其脱颗粒现象。有学者用直接免疫荧光观察,发现增生的小血管周围有IgA、IgM及补体C3沉积。还有学者在KD患者的血清中发现有白色念珠菌抗体,故推断白色念珠菌可能是致敏原。呼云之等〔2〕总结了37例KD病例,发现从事工农业劳动者居多,认为与患者的工作生活环境及个人卫生习惯有关,可能是由慢性感染所致的良性增生。沈明等〔3〕报道的7例多为个人卫生欠佳而健康状况良好者。
2流行病学特点
KD发病有明显的区域特点,绝大多数发生于东亚和东南亚,如:中国、日本、新加坡、印尼等地。欧美虽有少数病例报道,但患者也多为亚裔。而在我国,又多见于河北、内蒙、新疆、湖南、湖北、广东、贵州、四川、江西等地,港台也有报道。
KD好发于青、中年男性,20~50岁者占70%以上。所报道的最小年龄为5岁,最大为80岁。男女之比为4~7∶1〔2〕。
3临床表现
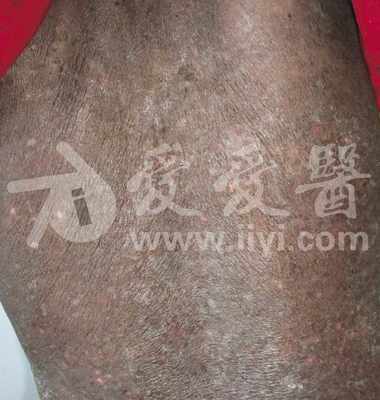
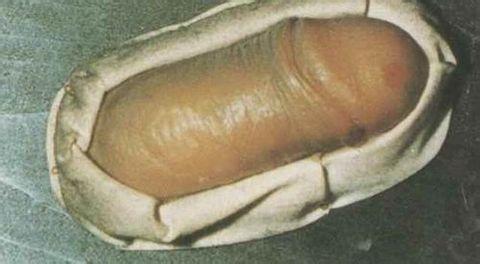
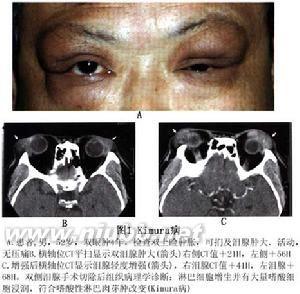
此病为起病缓慢、病程漫长(数年~十余年)的良性疾病。①软组织多发性(很少有单发)肿块是最常见的临床表现,肿块可同时或先后出现,75%位于头颈部的颌面区,多见于耳周、腮腺、颌下及颊部等处,有时也可累及唇、鼻背、鼻前庭、内眦、眶内、泪腺、颞部、枕后和悬雍垂、软腭、鼻咽等部位〔4〕。身体其它部位受累的有:肩背部、乳腺、胸壁、臀部、脾脏及外生殖器、正中神经等。肿块特点:边界不清,无痛,与皮肤粘连,活动度差。大小多为1~10cm直径,融合成团块者可超过10cm以上。早期质地似软橡皮,随病程延长逐渐变硬韧。肿块增长缓慢,可多年无明显变化。②淋巴结肿大也是很常见的临床表现,多见于颏下、颌下、颈部之浅表淋巴结。常多个淋巴结受累,有时也可累及腹股沟、腋下及肺门淋巴结。③皮肤瘙痒及色素沉着,发生率为40%~100%。多发生于肿块处的皮肤,可有斑点状皮疹和渗出,严重者局部糜烂、溃破。也有全身瘙痒者。
4实验室检查
①KD主要的实验室检查特点为外周血象中嗜酸性粒细胞比例和计数明显升高,比例多为10%~20%,最高达69%,直接计数达9.4×109/L。尚有不少报道白细胞总数也升高者。宫恩甲〔4〕发现以上指标与肿块的消长基本呈动态平行关系,并先于肿块复发前上升,故提出将上述指标的检测作为临床观察疗效、估计预后的客观指标。②血清IgE升高。③骨髓穿刺发现骨髓象中嗜酸性粒细胞也明显升高,主要为晚幼和成熟阶段〔2,4〕。④影像学检查无特异性,CT和MRI不易将此病与恶性肿瘤、淋巴瘤及血管瘤相鉴别〔5〕。
5病理表现
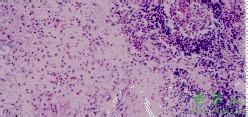
肿块无被膜,与周围组织无明显界限,可向周围组织、器官内浸润性生长。镜下见毛细血管大量增生,血管内皮细胞肿胀并明显增生,致管壁增厚甚至管腔阻塞。血管内皮增生区内有大量的淋巴细胞和嗜酸性粒细胞浸润,淋巴滤泡形成,嗜酸性粒细胞密集形成局限性的“嗜酸性小脓肿”灶。此外尚有不同程度的组织细胞、肥大细胞、浆细胞浸润。受累的淋巴结内淋巴滤泡增生活跃,生发中心扩大,嗜酸性粒细胞浸润于皮质、髓质及被膜下,严重者淋巴结结构消失。不同程度增生的纤维结缔组织包绕、分隔病变组织〔1,2〕。
6并发症
约有12%的KD患者伴有肾病综合症〔6〕,少数伴有支气管哮喘。
7诊断
临床上如遇头颈部无痛性肿块伴皮肤瘙痒,病程漫长,外周血象嗜酸性粒细胞及血清IgE升高者,应高度怀疑KD。确诊需依赖病理活检。细针穿刺细胞学检查(FNA)结果不可靠,不能作为诊断的常规手段。而对复发病例,因可避免重复活检,FNA是有价值的。
8鉴别诊断
本病易误诊为颈淋巴结结核、恶性淋巴瘤、纤维瘤、混合瘤等,病理可以确诊。
KD与血管淋巴样增生伴嗜酸性粒细胞增多症(ALHE)的关系:长期以来,对此两种疾病间的关系的争议很大。ALHE由Wells于1969年首先发现,多由西方国家报道。因其临床表现及病理改变与KD相似,故不少学者将两种疾病混为一谈,或认为是同一种疾病的不同阶段。但更多的学者认为二者为不同的、相互独立的疾病,其依据为:ALHE患者多为非东方人群,以中年女性多见。病变部位表浅,很少发生于腮腺及其他深部组织。肿块直径多小于2.0cm,边界清晰。病理改变与KD相似,但以血管内皮增生为主,淋巴细胞及嗜酸性粒细胞浸润但无淋巴滤泡形成,常表现为较大的动脉或静脉被其周围片状增生的小血管或毛细血管网所包围,血管内皮细胞呈异型性增生,指状突向管腔〔7,8〕。Olsen等〔9〕分析了116例ALHE病例后提出:ALHE应为包括KD、假性或非典型性化脓性肉芽肿、组织细胞样血管瘤、上皮样血管瘤等在内的、具有血管异常增生病理表现的一系列疾病。
9治疗
本病对放疗敏感,多家报道有效率为100%,是共认的首选治疗方法。杨洋等〔1〕发现放疗后病灶内毛细血管内皮转化为正常,嗜酸性粒细胞消失,淋巴细胞显著减少。照射剂量一般为20~30Gy即可〔10,11〕,无需照射至肿块完全消退,因放疗结束后未完全消退的病灶可继续缩小。柳文斌〔11〕认为只要肿块缩小70%以上即可停照,以减少不必要的照射反应。对于放疗后复发的病灶,再次放疗仍有效〔4〕。
用化疗治疗KD的报道较少。虽然口服强的松30~60mg/d能使肿块明显缩小或消失〔12〕,但停药后极易复发〔13,14〕,故不宜作为唯一的治疗手段。强的松治疗后复发者,放疗效果良好,目前强的松主要用于当KD并发肾病综合征时。CTX、COFP等化疗对KD无效〔1〕。
因肿瘤边界不清,手术不易彻底切除。单发肿块可考虑采用手术切除,而对多发者可手术结合放疗,采取切大放小、先放后切的联合治疗,否则极易复发〔1,2〕。
10预后
本病为良性病变,预后良好,但较易复发。除肾病综合征及支气管哮喘外尚未发现有其它重要脏器的并发症。至今无因KD而死亡的病例报道。田俊芝等〔10〕认为对治疗后复发者可长期观察,不必急于放疗或手术,当肿块增长迅速或较大而影响功能时再采取治疗。周青等〔7〕从KD患者的病理中淋巴细胞形态变化和免疫组化结果发现本病具有从良性向恶性发展的渐进性过程。宫恩甲〔4〕曾报告2例KD合并恶性淋巴瘤,其中1例死亡。